
Gene Therapy – FDA Takes Steps Toward Clarifying Scope of Orphan Drug Exclusivity
by Emily Marden and Anna Sims 
Gene therapy is the focus of concentrated industry attention, with numerous products in development to treat a variety of serious chronic diseases.1 The U.S. Food and Drug Administration (FDA or the agency) is taking active steps to facilitate this development. In July 2018 alone, the agency issued six draft guidance documents on gene therapy.2 In addition, in August 2018, FDA and the National Institutes of Health (NIH) announced efforts to streamline review of gene therapy protocols by, among other things, eliminating the regular required review of every protocol by the NIH Recombinant Advisory Committee (RAC), a federal advisory committee established in 1974 to advise on research related to the manipulation of nucleic acids.3
While FDA’s focus on gene therapy has resulted in a spate of useful guidance and policy statements, there remain critical questions about the applicability of certain existing programs, including the scope of available orphan drug exclusivity. Orphan drug exclusivity has been the subject of extensive regulatory attention since the passage of the Orphan Drug Act (ODA) in 1983. However, FDA has not issued regulations or guidance clarifying how the agency will interpret the ODA as it applies to gene therapy products. The key issue is whether FDA will characterize a gene therapy product for purposes of orphan drug exclusivity as the transgene component of the product alone, or alternatively, as the transgene plus any delivery vehicle (e.g., virus).
FDA now appears to be clarifying its approach. In at least two recent presentations by Acting Directors of FDA’s Office of Orphan Products Development (OOPD), FDA has indicated that it takes a transgene-plus-delivery-vehicle approach, subject—on a case-by-case basis—to ODA policy considerations. FDA’s stance on this issue is critical in determining the difficulty of obtaining orphan drug exclusivity and the timing of approval for follow-on gene therapy products.
Gene Therapy
Gene therapy products are regulated under the Federal Food, Drug, and Cosmetic Act (FDCA) as biological products.4 FDA has defined these products to include “all products that mediate their effects by transcription or translation of transferred genetic material, or by specifically altering host (human) genetic sequences.”5 According to FDA, these products can include nucleic acids, genetically modified microorganisms, engineered site-specific nucleases used for human genome editing, and ex vivo genetically modified human cells. Gene therapy products use physical, chemical, or viral delivery methods to deliver genetic material to a patient directly or to cells that have been extracted from a patient with the intention of reintroducing the modified cells back into the patient.
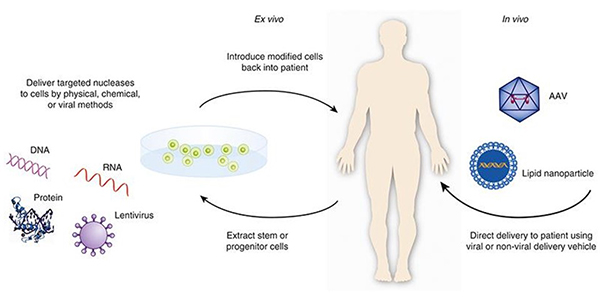
After an extended pause in the development of gene therapy products following a high profile clinical trial death in 1999,6 FDA approved Kymriah (tisagenlecleucel) in August 2017. Kymriah is a chimera antigen receptor T-cell (CAR-T) therapy intended to treat certain children and young adults with B-cell acute lymphoblastic leukemia. The agency has since approved two other gene-therapy products, Yescarta (axicabtagene ciloleucel) and Luxturna (voretigene neparvovec-rzyl). Both Kymriah and Yescarta received orphan drug exclusivity. According to an FDA official, around 70 percent of gene therapy Investigational New Drug Applications (INDS) are for rare diseases, and there has been a rapid increase in requests for orphan drug designations for gene therapy products over the last few years.7 FDA’s orphan drug designation database indicates that there are multiple such designations for gene therapies within single disease categories (e.g., hemophilia A, hemophilia B, Duchenne muscular dystrophy).
Orphan Drug Exclusivity
The ODA offers seven years of exclusive marketing rights for approval of an appropriately designated orphan drug or biological product. A sponsor seeking orphan designation for a drug must submit a request for designation to OOPD with the information required in 21 C.F.R. § 316.20 (content and format of a request for orphan-drug designation) and § 316.21 (verification of orphan-drug status). Orphan drugs and biological products are defined as those intended for the safe and effective treatment, diagnosis, or prevention of rare diseases/disorders that affect fewer than 200,000 people in the U.S., or that affect more than 200,000 persons but are not expected to recover the costs of developing and marketing a treatment drug.8
During the seven-year exclusivity period, FDA may not approve another version of same therapeutic product for the same disease or condition, subject to certain exceptions described below, even if the sponsor of the subsequent product submits a full New Drug Application (NDA) or Biologics License Application (BLA).9
The policy goal of the ODA is clear: a sponsor receives exclusivity as an incentive for developing therapeutic products for diseases and conditions affecting small populations. In addition to the seven years of exclusivity, products receiving an orphan drug designation are eligible for certain financial incentives, including tax credits up to 25 percent for qualified clinical trial costs and a waiver of FDA user fees.
Importantly, the scope of orphan drug exclusivity is limited to ensure maximum benefits to patients. Orphan drug exclusivity does not block FDA approval of the same therapeutic product for a different disease, nor does it block approval of a different therapeutic product for same disease. Moreover, there are three statutory exceptions that permit FDA to approve the same therapeutic product for the same disease or condition in spite of existing orphan drug exclusivity. These include: (1) if the holder of orphan drug exclusivity cannot “ensure the availability of sufficient quantities of the drug to meet” patient needs; (2) if the holder of the orphan drug exclusivity consents; or (3) if the subsequent product is “clinically superior to any already approved or licensed drug that is the same drug.”10 The third exception is important: FDA can approve a product that is “the same” as a previously approved orphan product if there is a demonstration that the second product is “clinically superior” to the first. Such approvals are relatively rare, and FDA is careful to limit such approvals to clear evidence of superiority.11
When is a Gene Therapy Product the “Same” for Purposes of Orphan Drug Exclusivity?
In the orphan drug context, the term “same” does not mean that two drugs must be “identical.” Rather, FDA has long taken the position that two drug products may be considered the “same” orphan drug product even if there are certain differences between the two products.12 At the same time, there is no clear FDA precedent for determining the sameness of a complex gene therapy product containing both genetic material and a delivery vehicle, such as a virus or other vector containing a transgene. FDA could characterize the product solely as the transgene, with the implication that any subsequent gene therapy product containing the same transgene cannot be approved for seven years absent a demonstration of clinical superiority, regardless of changes to the delivery vehicle. Alternatively, the agency could apply the orphan drug exclusivity to the transgene plus delivery vehicle, opening the door to approval of subsequent products utilizing different vectors within the seven-year window. These alternate approaches could influence how difficult it is to obtain orphan drug exclusivity, thus changing development incentives for sponsors, market competition, and availability of treatment options for patients.
Existing regulations reflect the importance of orphan drug policy considerations in making sameness determinations for therapeutic products. The sameness of a small molecule drug is established by comparing the active moiety of two products, but for macromolecules, “same drug” means that it contains the “same principal molecular structural features (but not necessarily all of the same structural features) and is intended for the same use as a previously approved drug.”13 In the 1992 Final Rule on Orphan Drug Exclusivity, FDA responded to a comment proposing that the concept of “active moiety” be applied to both micro and macromolecular products. FDA disagreed, explaining that for small molecule products, “any change in covalent structure creates a new active moiety whose properties may well differ from the old active, moiety;” conversely, for macromolecules,
it would be trivially easy to make minor covalent changes that would leave the activity of the drug unaltered, but would create a “different drug” if the micromolecular definition of active moiety were to be used. This would render exclusive marketing of macromolecular drugs meaningless and would decrease incentives to develop orphan drugs. When such a change is meaningful, of course, it deserves, and under the rule would gain, exclusive marketing.14
The net result is that FDA retains broad discretion in identifying the relevant portion of complex products for purposes of identifying the “same drug.”
Emerging FDA Position
Recently, OOPD leadership has offered key insights into how FDA may characterize gene-therapy products for purposes of exclusivity and hinted at forthcoming guidance on the topic. Comments by the then Acting Director of OOPD at a May 4, 2018 Food and Drug Law Institute conference point to the agency’s likely position. With regard to sameness determinations, she stated that for “totally different vectors . . . [FDA] would look at that as different. Similarly, different transgenes, we would look at that as different. But when you get to serotypes or promoters, we really have to go back and communicate.”15 In other words, FDA appears to consider the product to be comprised of transgene-plus-vector for purposes of sameness determinations. Thus, FDA would consider products with the same transgene but different vectors to be different drugs, and not blocked by exclusivity. However, the statement suggests that changes to a vector—such as serotypes or promoter regions—may not qualify a product as a different drug subject to policy considerations.
Subsequently, on August 20–21, 2018, the NIH National Center for Advancing Translational Sciences (NCATS) and the FDA Center for Biologics Evaluation and Research (CBER) hosted “The Growing Promise of Gene Therapy Approaches to Rare Diseases” workshop. At this event, a former Acting Director of OOPD reiterated that in the absence of regulations or applicable precedent, FDA would take a case-by-case approach to exclusivity in the context of the policy aims of the ODA, namely encouraging innovation while ensuring the best available treatments for patients.16 The presentation focused on key product features FDA would consider in determining when a gene therapy product was “the same.” These include the vector, the transgene, other regulatory elements of transduction and expression, and manufacturing issues, such as those related to target cells. This former Acting Director noted that a difference in manufacturing would not de facto make a gene therapy product different than a predecessor—a comment that echoes certain positions taken by FDA in the context of follow-on complex drugs.
Looking Ahead: Ongoing Need for Clarity
FDA comments on the characterization of gene therapy products for purposes of orphan drug exclusivity are welcome for an industry waiting for clear signals from the agency and will help in strategic planning around product development and market expectations. At the same time, other key questions remain, including what the specific criteria are for identifying a plausible hypothesis of “clinical superiority” in the gene therapy context, and the scope of information necessary to demonstrate such clinical superiority. FDA has previously suggested that it might offer guidance on these issues specifically for complex biological products, such as plasma protein therapies.17 However, to date the agency has not done so. If the recent flurry of agency activity on gene therapy is any indication, it is reasonable to be cautiously optimistic that FDA may offer further guidance on this complex and evolving area.
Update Magazine
December 2018/January 2019




- FDA currently has more than 700 active investigational new drug applications for gene therapies. F. Collins and S. Gottlieb, The Next Phase of Human Gene-Therapy Oversight, NEJM (Aug. 15, 2018), https://bit.ly/2wctXmJ.
- FDA Statement, Statement from FDA Commissioner Scott Gottlieb, M.D. on agency’s efforts to advance development of gene therapies (July 11, 2018), https://bit.ly/2NK6pgH.
- F. Collins and S. Gottlieb, supra note 1.
- See Section 351 of the Public Health Services Act (PHSA).
- Draft Guidance, Long Term Follow-Up [“LTFU”] After Administration of Human Gene Therapy Products at 28 (July 2018), https://bit.ly/2O4C3VE; see also 58 Fed. Reg. 53,248, 53,249 (Oct. 14, 1993); FDA Guidance, Guidance for Human Somatic Cell Therapy and Gene Therapy at 3 (Mar. 1998), https://bit.ly/2ONy0OE.
- S. Stolberg, The Biotech Death of Jesse Gelsinger, NYT (Nov. 28, 1999), https://nyti.ms/2Mi5Rxl.
- I. Irony, Director, Division of Clinical Evaluation and Pharmacology/Toxicology, Office of Tissues and Advanced Therapies, CBER, The Growing Promise of Gene Therapy Approaches to Rare Disease (Aug. 21, 2018).
- FDA, Developing Products for Rare Diseases & Conditions, https://bit.ly/2MTjp27; 21 C.F.R. § 316.20(b)(8).
- Section 527(a) of the FDCA.
- Section 527(b) & (c) of the FDCA.
- Only one product is currently listed on FDA’s website containing post-2017 summaries of clinical superiority findings. See FDA, Clinical Superiority Findings, https://bit.ly/2NTf1p5; see also Section 527(e)(2) to the FDCA.
- See, e.g., I. Irony, supra note 7.
- 21 C.F.R. § 316.3(b)(ii).
- 57 Fed. Reg. 62,075, 62,077 (Dec. 29, 1992).
- D. Gingery, US FDA’s Acting Orphan Products Development Director Has Gene Therapy Background, The Pink Sheet (May 6, 2018).
- I. Irony, supra note 7.
- 78 Fed. Reg. 35,117, 35,122 (June 12, 2013).
