Advancing the Transition to Computer Software Assurance:
Responding to the FDA Draft Guidance for Production and Quality System Software
Introduction
Life sciences companies increasingly rely on computerized systems within manufacturing and quality, which can include automation, robotics, simulation, and other digital capabilities. These technologies provide significant benefits for enhancing the quality, availability, and safety of medical devices.[1] Over the past decade, most life sciences organizations have transitioned their regulated systems into the cloud. This is reflected by a global market for cloud services in the medical device sector of $2 billion in 2021, forecast to increase by a compound annual growth rate of 17.1% to $4.4 billion by 2024.[2]
Under FDA regulations covering Good Manufacturing Practice and related best practices (known collectively as GxP), it is necessary to validate systems with direct or indirect product and patient impact prior to using them in a production environment. These types of “GxP-facing” systems can include quality management systems, electronic document management systems, batch record systems, laboratory information management systems, clinical systems, product safety and complaint reporting, environmental monitoring, and systems for transferring and analyzing production data.[3] Since these systems form the technological backbone of life sciences companies, computer system validation (CSV) is a highly regulated and required activity for life science companies. The burden placed on organizations to manage CSV has increased with the proliferation of specialized computerized systems and is further amplified by a global staffing shortage. Based on stakeholder feedback, the U.S. Food and Drug Administration (FDA) is now encouraging a transition to computer software assurance (CSA) to support data integrity, product quality, product safety, and patient safety.[4] This transition marks an evolution in validation effort from documenting extensive testing to focusing on critical thinking and risk management up-front.[5] CSA offers a path away from time- and personnel-intensive, burdensome validation to a risk-based, workflow-driven process that preserves data integrity.[6]
On September 13, 2022, the FDA Center for Devices and Radiological Health (CDRH) and the Center for Biologics Evaluation and Research (CBER) issued a long-awaited document titled, “Computer Software Assurance for Production and Quality System Software: Draft Guidance for Industry and Food and Drug Administration Staff.”[7] The draft guidance responds to stakeholder requests for greater clarity on FDA’s expectations for software validation for computers and automated data processing systems and for a more agile, iterative approach to the validation of software used in these areas.[8] FDA “believes that these recommendations will help foster the adoption and use of innovative technologies that promote patient access to high-quality medical devices and help manufacturers to keep pace with the dynamic, rapidly changing technology landscape, while promoting compliance with laws and regulations implemented by FDA.”[9]
Despite the fact that this is draft guidance, FDA and key industry leaders have maintained that risk-based, lean validation approaches are acceptable, and that companies “have the flexibility they need to adjust the extent and stringency of controls based on any factors they choose.”[10] CSA falls within this category, meaning that companies can safely adopt CSA approaches. However, to date, adoption has been slow, largely because of concerns about potential inspection risks due to changes in the approach and potential changes in the validation deliverables required, coupled with uncertainty about how to apply critical thinking to validation.
This article provides a regulatory overview and next steps for companies, including key features of the new draft guidance; a comparison between CSV and CSA; suggestions on overcoming reluctance to adopt CSA concepts; a discussion of how to align validation testing with level of risk; ideas on how to move towards inspection readiness with CSA; and potential gaps in the guidance that stand in the way of acceptance and implementation of the CSA guidance by industry.
Regulatory Overview
The September 2022 draft guidance describes CSA as “a risk-based approach to establish confidence in the automation used for production or quality systems,” based on a four-step process. This involves identifying the intended use of the software within the production and quality systems, determining a risk-based approach depending on possible outcomes if the software did not perform as intended, determining appropriate assurance activities based on that risk, and establishing an appropriate record with enough evidence to show that the software was assessed and performs as intended (Figure 1).[11]
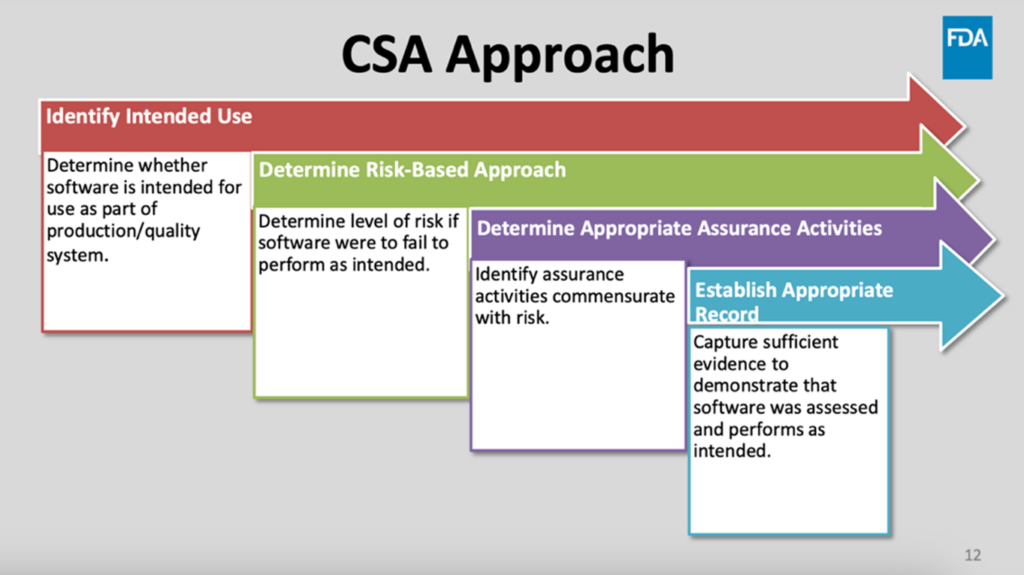
Figure 1: FDA’s explanation of the computer software assurance (CSA) approach[12]
|
Sidebar: FDA Definition: Computer Software Assurance “Computer software assurance [CSA] is a risk-based approach for establishing and maintaining confidence that software is fit for its intended use. This approach considers the risk of compromised safety and/or quality of the device (should the software fail to perform as intended) to determine the level of assurance effort and activities appropriate to establish confidence in the software. . . . Such an approach supports the efficient use of resources, in turn promoting product quality.” U.S. Food and Drug Administration Draft Guidance on “Computer Software Assurance for Production and Quality System Software;” September 13, 2022[13] “FDA is issuing this draft guidance to provide recommendations on computer software assurance for computers and automated data processing systems used as part of medical device production or the quality system. FDA believes that these recommendations will help foster the adoption and use of innovative technologies that promote patient access to high-quality medical devices and help manufacturers to keep pace with the dynamic, rapidly changing technology landscape, while promoting compliance with laws and regulations implemented by FDA. This draft guidance is not final nor is it for implementation at this time.” Federal Register Notice on Draft CSA Guidance; September 13, 2022[14]
|
|
Sidebar: An FDA Description of CSA Computer software assurance has the following features:
FDA webinar, “Draft Guidance on Computer Software Assurance for Production and Quality System Software;” October 27, 2022[15] |
The draft guidance applies to assurance activities for computer systems and automated data processing systems used as part of medical device production or the quality system, including software for design, development, manufacturing, or the quality system.[16] The document does not apply to software used as a medical device (SaMD) or software in a medical device (SiMD).[17]
Software validation has traditionally involved software testing and verification at every stage of software development, along with exhaustive documentation. According to FDA, software testing alone is often insufficient to establish confidence that the software is fit for its intended use: “FDA believes that applying a risk-based approach to computer software used as part of production or the quality system would better focus manufacturers’ assurance activities to help ensure product quality while helping to fulfill the validation requirements of Title 21 of the Code of Federal Regulations or CFR Part 820.70(i).”[18]
|
Sidebar: Key Features of the September 2022 Draft Guidance
|
The CSA draft guidance builds on the agency’s framework for computer system validation issued in 1997 with 21 C.F.R. Part 11.[19] This was later refined in “General Principles of Software Validation: Guidance for Industry and FDA Staff,” published in 2002.[20] The draft guidance aligns with multiple recent initiatives, including the FDA Case for Quality program and Advanced Manufacturing efforts,[21] Industry 4.0/Pharma 4.0/Validation 4.0, and the release of the second edition of the International Society for Pharmaceutical Engineering (ISPE) Good Automated Manufacturing Practice 5 (GAMP 5).[22] These publications indicate that regulatory bodies favor a risk-based approach to assure the data integrity and suitability of computer systems to facilitate the speed of innovation, manufacturing, and product delivery.[23]
Computer Systems Validation (CSV) v. Computer Software Assurance (CSA)
Traditional CSV is a burdensome, often paper-based, Stage-and-Gate process that involves a blanket approach to validation testing of systems, features, and changes without regard to risk. Life sciences and medical device manufacturers that are covered by 21 C.F.R. Part 820 (Quality System Regulation) have historically carried out massive validation and verification testing efforts—often recorded as vast numbers of screenshots—in the name of compliance with regulations.[24] Such efforts, however, may not appropriately test whether a system is fit for the intended use, potentially resulting in risks to product quality and patient safety. Additionally, since traditional validation is time- and resource-intensive, companies often fall behind on validating the scheduled upgrades that come with Software-as-a-Service (SaaS) applications. This can cause them to fall out of compliance and into “technical debt,” because they are running on outdated or insufficiently validated systems. If we consider validation activities in the context of the Pareto Principle (or the “80-20” rule), CSV typically comprises 80% effort on documentation and testing and only 20% focus on critical thinking and assurance activities.[25] CSA increases efficiency by reversing these percentages, with an 80% focus on critical thinking and 20% of effort going into documentation and testing (Figure 2).[26] This change in structure of effort can provide significant time and cost savings in the assurance activities (validation) of GxP-facing applications, while promoting a path out of technical debt and into better compliance.
Figure 2: Computer systems validation (CSV) versus computer software assurance (CSA)
Historical factors contribute to making traditional computer systems validation problematic:
- Misunderstanding of the roots of the need to validate: The original directive to validate comes from two primary sources, 21 F.R. Part 11 (intended to ensure that electronic signatures and records are as robust and reliable as their paper counterparts) and 21 C.F.R. Part 820, the Quality Systems Regulation (in particular, 21 C.F.R. Part 820, Subpart G).[27] Subpart G was initially meant to focus on the underlying processes (such as manufacturing) but was also applied for the purpose of computer systems validation. At the time, the use of computer systems in the life sciences industry was new, and technology was often homegrown due to the lack of off-the-shelf alternatives. IT and QA executives knew they had to test their computer systems, but computer validation could not necessarily be done in the same manner as process validation. This caused confusion around the best ways to test and the extent of testing required to ensure that a computerized system was fit for intended use. The result was a default to a blanket, test-everything approach, laden with screenshots.[28]
- Home-grown, custom-built systems were the norm for life sciences companies in the 1990s, since there were few, if any, commercial options for these specialized systems. Companies often had large software development and CSV departments that built, tested, updated, and retested homegrown systems that resided on local servers—a model that was slow, costly, and difficult to scale. In the 2000s, large-scale development of commercial SaaS systems enabled companies to quickly scale applications and services. Although traditional CSV was a time-consuming barrier that often resulted in technical debt, companies adhered to the CSV approach in the absence of alternatives.
Taking a blanket approach to testing may have been appropriate in the 1990s, when software development was nascent. Since then, there have been major advances in software design and development, as well as in the tools used by software engineers to develop and test systems. The Stage-and-Gate or Waterfall approach commonly used in the late 1990s (Figure 2) aligned well with traditional CSV, but it has not maintained that alignment as technology evolved. The shift to the Agile development methodology, with teams working in sprints and testing iteratively throughout development, meant that systems could be developed and improved with greater speed and efficiency. Traditional CSV cannot keep pace with Agile methodology, posing challenges to regulated companies. ISPE’s GAMP 5 was rolled out in 2008 to help bridge this gap and support compliance, and a second edition of GAMP 5 was released in July 2022 to account for advancements in technology.[29] CSA aligns with and expands upon GAMP 5 to provide the life sciences industry with an updated explanation of FDA’s thinking around risk-based, iterative, Agile-compatible approach to assuring the quality of computerized systems.
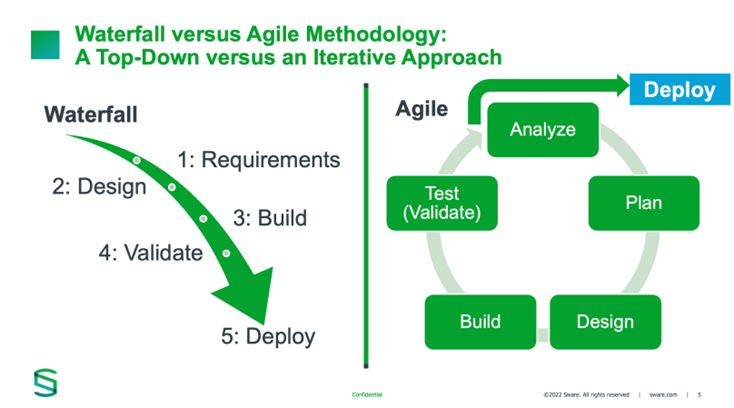
Figure 2: Waterfall versus agile methodology
As technology advanced in the 2000s and cloud computing became the norm, regulations fell behind. While new, cost-effective, scalable applications were available, companies were still using traditional paper-based methods to document and test every feature of every software application being released into production—whether for an initial implementation, upgrade, or change. Compliance with regulations, including 21 C.F.R. Part 820 and 21 C.F.R. Part 11, took priority over the fitness-for-intended use of the software solutions. The reason for this was a perceived concern that if companies departed from the traditional approach, inspectors would find fault with their CSV deliverables, risking poor audit outcomes, Form FDA 483 observations, and/or Warning Letters.[30]
|
Sidebar: Features of Traditional CSV The traditional CSV approach: 1) Does not set the level of testing based on risk; 2) Takes a reactive, “audit proofing” approach, as opposed to a proactive, risk-based approach; 3) Generates large, burdensome quantities of paper documents; 4) Is often encumbered by test script errors and large numbers of screenshots; and 5) Involves performing unnecessary activities in an effort to comply with regulations but may not appropriately test the system or its state of validation. |
Overcoming Reluctance to Adopt CSA Concepts
The concepts promoted in the CSA draft guidance and similar publications have been slow to gain traction in the medical device industry as well as across other regulated sectors of the life science industry that validate their systems, including pharmaceutical and biotech companies and manufacturers of active pharmaceutical ingredients (APIs). The CSA guidance itself is not a prescriptive document, but rather a reflection of FDA’s current thinking on the topic of computer software assurance. As such, the document does not prescribe specific “how-to” approaches for risk assessment, selection, and construction of validation deliverables, nor does it provide a specific measure of how much testing is enough to satisfy an inspection. This has been a stumbling block in gaining acceptance and promoting adoption of the CSA guidance throughout the life sciences industry. Companies may be concerned that using risk-based, less documentation-intensive, more lightweight approaches to validation and verification may result in inspection findings. There is also some confusion around the meaning and applications of critical thinking—not only for the validation testing process itself, but for elements such as vendor qualification (a key to leveraging vendor validation deliverables), assigning appropriate types of testing (especially unscripted and ad hoc testing), and determining the appropriate amount of documentation.
|
Sidebar: Demystifying the Application of Critical Thinking to Validation The term “critical thinking” has become ubiquitous when discussing CSA, even though the term does not appear in the guidance itself. What is critical thinking, and why is it important? According to Shitamoto and Gurumoorthi (2021), “CSA is the application of critical thinking to validation that adds risk-based documentation to risk-based testing while taking a lifecycle approach, to ‘take credit’ for activities, and reduce the validation effort.”[31] In addition to employing a risk-based approach, critical thinking involves:
|
As technology advances and hosted systems (such as SaaS) are increasingly adopted, companies are experiencing challenges in keeping systems in a validated state (also known as “a state of control”). The effort and resources required can be overwhelming—especially considering periodic system upgrades, patches, and interim releases, all of which must be validated prior to being moved into production. Companies that avoid risk-based approaches such as CSA and GAMP 5 run the risk of falling behind and out of compliance.[33]
An approach that considers impact on product safety, product quality, and patient safety will help companies stay abreast of technological change. Knowing which features are most critical, most complex, and highest risk will enable companies to concentrate testing on those features. Software vendors frequently offer customers access to validation documentation, which means that companies can avoid repeating tests already carried out by the vendor. The ability to leverage the validation deliverables of potential software vendors is an important additional factor in determining the risk of a system. Vendors should be vetted to determine the robustness of their quality system, the quality and integrity of their software products, and the strength of their validation documentation. A supplier audit or other form of assessment enables companies to determine whether they will be able to qualify the vendor, and thereby leverage the vendor’s systems, services, and/or validation deliverables.
Aligning Validation Testing with Level of Risk
Risk—the amount of potential harm to patient safety, product safety, and/or product quality; the likelihood of such harm; and its detectability—is a key element in CSA. In traditional CSV, the customer subsumed all the risk of validating a purchased system, which meant complete retesting of applications to ensure fitness for the intended use. With the CSA approach, the risk is distributed between the customer and the vendor. Enabling the use of vendor-produced deliverables (via supplier qualification activities executed by the customer) avoids the repetition of testing and facilitates compliance with regulatory requirements under 21 C.F.R. Part 820. Risk-related activities should include impact assessments of the potential for the system to malfunction and the extent to which key processes impacting data integrity, data availability, data security, product quality, product safety, and patient safety would be affected.
A thorough risk-impact assessment should be carried out at the beginning of each project to determine potential risks/hazards surrounding the application, and the criticality of the features or requirements to the business process. Risk reviews should be executed periodically throughout the project to ensure that risks are being appropriately mitigated. Risk assessments should be executed consistently and objectively, using standardized, documented risk impact assessment questionnaires with built-in numeric scales. This approach provides a demonstrable assessment of risk, including quantifiable data to support the assessment. Activities can then be planned to ensure that risks are mitigated appropriately.
Rigor and type of testing required are determined based on the overall risk. As a general rule, scripted testing is used for applications that are complex, have features with direct impact on patients and products, and pose the highest levels of risk. Unscripted and ad-hoc testing are used where risk is lower. CSA provides the flexibility to select the testing methodology that suits the size, complexity, and risk of the application. The testing methodologies chosen should be explained in the validation plan and include the results in the validation test summary report.
There are validation automation platforms on the market that include tools to promote the quantifiable and objective assessment of risks, making it straightforward to assess risk initially and to apply the same approach during consequent risk reviews. These platforms also offer the advantage of automating validation protocol workflows, testing, and even reporting and dashboards. As with GAMP 5 (second edition), the CSA draft guidance indicates that it is acceptable to use such tools as systems of record for validation activities.
Moving Towards Inspection Readiness with CSA
FDA recommends that CSA records “retain sufficient details of the assurance activity to serve as a baseline for improvements or as a reference point if issues occur.” Documentation of assurance activities “need not include more evidence than necessary to show that the software feature, function, or operation performs as intended for the risk identified.”[34]
Based on examples in the draft guidance, the types of deliverables remain the same but may differ slightly from project to project. The size of the documents will likely be smaller than with traditional CSV activities. Cornerstone documents generally remain for most projects, including:
- The validation plan that explains strategy and thinking
- The risk assessment (and results of any risk reviews)
- Test objectives and results of test protocols (depending on the system, these may include installation, operational, performance, and user acceptance testing)
- Records indicating who performed the testing and when
- A summary report of the testing performed and the results, any software issues found and their resolutions, conclusions reached as a result of the testing, and a statement as to the suitability of the system for use in production
Experts responsible for approvals are likely to remain largely the same; however, they may come into the picture in a different order in CSA compared to CSV. In CSA, the Quality function (such as QA-IT) is involved earlier in the project for critical thinking and planning and remains active during risk reviews throughout the project. By contrast, traditional CSV often left QA out until project end, resulting in significant rework and backtracking due to risks and deficits that could have been caught earlier, had Quality experts been made aware.
FDA encourages the use of automated tools for testing, traceability, and electronic capture of test results, as opposed to a paper-based approach. The agency also recommends the use of electronic records, such as system logs, audit trails, and other data generated by the software, as opposed to paper documentation and screenshots, in establishing the record associated with the assurance activities.
The automation focus is consistent with the approach recommended in the second edition of GAMP 5. Automation provides the advantage of ensuring that records of assurance activities are stored in a single location, as a single source of truth, thus preventing errors or discrepancies in transcribing or copying validation collateral packages. Purpose-built validation automation platforms (VAPs) are available to automate these activities. VAPs also provide signature approval capabilities, enabling companies to use them to record validation deliverables, removing the need to cut, copy, and paste this information into static documents. VAPs also enable control of the information that is given to an inspector. Most VAPs include portals that allow inspectors to see the validation deliverables in the same system where they were created, making it easy to prepare for inspections, and provide inspectors with trust through transparency.
Conclusion
Historically, CSV activities resulted in burdensome documentation that may not have adequately tested a system’s fitness for its intended use. The updated CSA approach requires companies to assess risks; to think more up-front and test less but more wisely; to bring Quality and IT compliance into the project earlier; to integrate test automation; to leverage work the vendor has already done to assure the quality of the application; and to “right size” evidence to show that the system is performing as per its intended use in a reliable, consistent manner that preserves data integrity. Device companies should consider combining this line of thinking with validation automation technology to streamline testing and the capture of validation data. Growth in the use of VAPs will provide built-in vehicles for validation workflow management that supports these initiatives and provide a valuable source of inspection readiness. The incorporation of VAPs into the assurance process will prove key in implementing CSA successfully and with confidence.
In sum, FDA would like to see the medical device industry advance its focus on device features, automation, and high-quality manufacturing practices that promote data integrity, product quality, and patient safety. The agency has written the CSA guidance to be flexible enough to permit companies to determine the best approach to advance these initiatives while maintaining software quality. FDA recognizes that traditional approaches to CSV have hindered such advancements. Life sciences companies validating their GxP-facing applications should consider the advantages of a move to CSA, even if they remain cautious—keeping in mind that inspectors may be as unwilling to review thousands of pages of test scripts and screen shots as companies are to generate them. Companies can move confidently to modernize validation knowing that FDA supports adoption of CSA (and its advantages, such as fewer screen shots) and that supportive validation technologies are both available and acceptable.
[1] Webinar Transcript, Computer Software Assurance for Production and Quality System Software—Draft Guidance, U.S. Food & Drug Admin., Ctr. for Devices & Radiological Health, Oct. 27, 2022, https://www.fda.gov/media/163299/download [hereinafter CDRH Webinar Transcript].
[2] Global Data Healthcare, Medical Device Companies Embrace Cloud Services, Medical Device Network, June 24, 2021, https://www.medicaldevice-network.com/comment/medical-device-companies-cloud-services/.
[3] Draft Guidance: Computer Software Assurance for Production and Quality System Software: Draft Guidance for Industry and Food and Drug Administration Staff, U.S. Food & Drug Admin., Sept. 13, 2022, https://www.fda.gov/media/161521/download [hereinafter FDA, CSA Draft Guidance].
[4] Final Guidance: Data Integrity and Compliance With Drug CGMP Questions and Answers: Guidance for Industry, U.S. Food & Drug Admin., Dec. 2018, https://www.fda.gov/media/119267/download; Federal Register Notice, Computer Software Assurance for Production and Quality System Software; Draft Guidance for Industry and Food and Drug Administration Staff; Availability, 87 Fed. Reg. 56059, Sept. 13, 2022, https://www.federalregister.gov/documents/2022/09/13/2022-19763/computer-software-assurance-for-production-and-quality-system-software-draft-guidance-for-industry [hereinafter CSA Fed. Reg. Notice].
[5] White Paper, Transitioning from CSV to CSA: Taking a More Risked-Based Approach, ISPE, Feb. 1, 2023, https://ispe.org/pharmaceutical-engineering/transitioning-csv-csa-taking-more-risked-based-approach.
[6] Bryan Ennis, Moving Beyond Paper-Based Validation: An Industry Transformation, Sware, 2022, https://8383502.fs1.hubspotusercontent-na1.net/hubfs/8383502/SWR-82-Beyond-Paper-based-Whitepaper.pdf?hsCtaTracking=de9e931d-c4ac-4dd0-9955-24193aca708d%7Cb160babe-1dd8-42af-a50a-a3114f943717.
[7] FDA, CSA Draft Guidance, supra note 3; CSA Fed. Reg. Notice, supra note 4.
[8] CDRH Webinar Transcript, supra note 1.
[9] Id.
[10] Ken Shitamoto & Senthil Gurumoorthi, Understanding FDA’s CSA Guidance in the Context of Current Regulations and GAMP, America Pharmaceutical Review, March 29 2021, https://www.americanpharmaceuticalreview.com/Featured-Articles/574659-Understanding-FDA-s-CSA-Guidance-in-the-Context-of-Current-Regulations-and-GAMP/.
[11] Id.
[12] Id.
[13] FDA, CSA Draft Guidance, supra note 3.
[14] CSA Fed. Reg. Notice, supra note 4.
[15] CDRH Webinar Transcript, supra note 1.
[16] Shitamoto & Gurumoorthi, supra note 10.
[17] CDRH Webinar Transcript, supra note 1.
[18] National Archives Code of Federal Regulations Title 21, chapter I, subchapter H, last amended on Apr. 14, 2023, https://www.ecfr.gov/current/title-21/chapter-I/subchapter-H/part-820 [hereinafter 21 C.F.R. Part 820]; General Principles of Software Validation: Guidance for Industry and FDA Staff, U.S. Food & Drug Admin., Jan. 2002, https://www.fda.gov/files/medical%20devices/published/General-Principles-of-Software-Validation—Final-Guidance-for-Industry-and-FDA-Staff.pdf.
[19] Federal Register notice Food and Drug Administration 21 C.F.R. Part 11 [Docket No. 92N–0251] RIN 0910–AA29 Electronic Records; Electronic Signatures. Mar. 20, 1997, https://www.govinfo.gov/content/pkg/FR-1997-03-20/pdf/97-6833.pdf.
[20] Worried About Computer Software Assurance? Relax, You’re Probably Already Doing It, FiercePharma sponsored content by ValGenesis, Mar. 7, 2022, https://www.fiercepharma.com/sponsored/worried-about-computer-software-assurance-relax-youre-probably-already-doing-it.
[21] Advanced Manufacturing: FDA News, Events, and Funding Opportunities Related to Advanced Manufacturing, U.S. Food & Drug Admin., current as of Mar. 31, 2023, https://www.fda.gov/emergency-preparedness-and-response/mcm-issues/advanced-manufacturing.
[22] Case for Quality, U.S. Food & Drug Admin., 2020, https://www.fda.gov/medical-devices/quality-and-compliance-medical-devices/case-quality; Chip Bennett, Hans Heesakkers, Stefan Horneborg, Gilad Langer, Line Lundsberg-Nielsen, Anthony J. Margetts & Fritz Röder, Industry Perspective: Validation 4.0 – Shifting Paradigms, ISPE Pharmaceutical Engineering, Nov.–Dec. 2020, https://ispe.org/pharmaceutical-engineering/november-december-2020/industry-perspective-validation-40-shifting; ISPE, GAMP 5: A Risk-Based Approach to Compliant GxP Computerized Systems (2d Ed., July 2022), https://ispe.org/publications/guidance-documents/gamp-5-guide-2nd-edition; Sion Wyn, Christopher J. Reid, Chris Clark, Michael L . Rutherford, Heather D. Watson, Lorrie L. Vuolo-Schuessler & Anthony D. Perez, Why ISPE GAMP® Supports the FDA CDRH: Case for Quality Program, ISPE Pharmaceutical Engineering, Pharmaceutical Engineering, Nov./Dec. 2019, https://ispe.org/pharmaceutical-engineering/why-ispe-gamp-supports-fda-cdrh-case-quality-program.
[23] Stephen M. Hahn & Colin Rom, Accelerating the Adoption of Advanced Manufacturing Technologies to Strengthen Our Public Health Infrastructure, U.S. Food & Drug Admin., Jan. 15, 2021, https://www.fda.gov/news-events/fda-voices/accelerating-adoption-advanced-manufacturing-technologies-strengthen-our-public-health#:~:text=The%20FDA%20and%20industry%20have,memorandum%20of%20understanding%20(MOU.
[24] Worried About Computer Software Assurance?, supra note 20.
[25] Mark Best & Duncan Neuhauser, Joseph Juran: Overcoming Resistance to Organisational Change, Quality & Safety Health Care, Nov. 2006 Oct;15(5):380–82. Doi: 10.1136/qshc.2006.020016. PMID: 17074878; PMCID: PMC2565827, https://www.researchgate.net/publication/6721371_Joseph_Juran_Overcoming_resistance_to_organisational_change.
[26] Mary Beth Walsh, Computer Software Assurance (CSA): The FDA’s New Approach to CSV, Kalleid Inc., July 15, 2021, https://kalleid.com/computer-software-assurance-csa-the-fdas-new-approach-to-csv/.
[27] Worried About Computer Software Assurance?, supra note 20.
[28] Computer Systems Assurance: Making the Transition from Computer Systems Validation, AssurX blog, (undated), https://www.assurx.com/computer-systems-assurance-making-the-transition-from-computer-systems-validation/.
[29] Hahn & Rom, supra note 23.
[30] FDA Form 483 Frequently Asked Questions, U.S. Food & Drug Admin., current as of Jan. 9, 2020, https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/inspection-references/fda-form-483-frequently-asked-questions; Warning Letters, U.S. Food & Drug Admin., current as of Apr. 18, 2023, https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-actions-and-activities/warning-letters.
[31] 21 C.F.R. Part 820, supra note 18.
[32] Advanced Manufacturing, supra note 21.
[33] Hahn & Rom, supra note 23.
[34] FDA, CSA Draft Guidance, supra note 3.
 JACQUELIN DAVIDSON, Head of Regulatory Intelligence and Innovation at Sware, Inc., brings over 25 years of experience in the life sciences industry. Davidson is a recognized thought leader in IT quality assurance, specializing in software lifecycle management, quality risk management, and software validation for the pharmaceutical and medical device sectors.
JACQUELIN DAVIDSON, Head of Regulatory Intelligence and Innovation at Sware, Inc., brings over 25 years of experience in the life sciences industry. Davidson is a recognized thought leader in IT quality assurance, specializing in software lifecycle management, quality risk management, and software validation for the pharmaceutical and medical device sectors.Update Magazine
Summer 2023





