Regulations of CAR T-Cell Therapies—The Past, Present, and Future
Introduction
The field of oncology has always spurred interest and motivated researchers to develop drugs to address the mortality and quality of life associated with cancers. Conducting clinical trials and obtaining robust results sufficient to support regulatory approval may take years. Since time is of the essence for patients with potentially fatal malignancies, FDA has created processes to aid in the review and approval of novel therapies. Regenerative Medicine Advanced Therapy (RMAT) designation and Breakthrough Therapy designation were created to identify therapies that, based upon preliminary clinical evidence, indicate the drug has the potential to address unmet medical needs for a disease or condition. These processes, however, create additional concerns for monitoring and evaluating long-term side effects and serious complications that may accompany the much-needed benefits and are challenging to address in fast-paced clinical trials. CAR T-cell therapy is one such therapy, which is currently being extensively studied. Its complexity as a biologic and its fairly new experimentation in humans makes the safety monitoring a challenge from a regulatory perspective.
What is Chimeric Antigen Receptor T (CAR T) Cell Therapy?
CAR T-cell therapy is novel immunotherapy in which the patient’s T-cells are isolated by apheresis and engineered to form chimeric antigen receptors, after which they are grown in the laboratory and infused into the patient. Chimeric antigen receptors have the unique ability to bind to the cancer-related proteins that serve as a mechanism to attack the tumor. CAR T-cell therapy is currently approved for the treatment of some types of cancer and is being studied for a wide variety of other oncological indications.[1]
To better understand CAR T therapy and its implications, it is helpful to delve into its history and the regulatory processes in place that oversee these therapies.
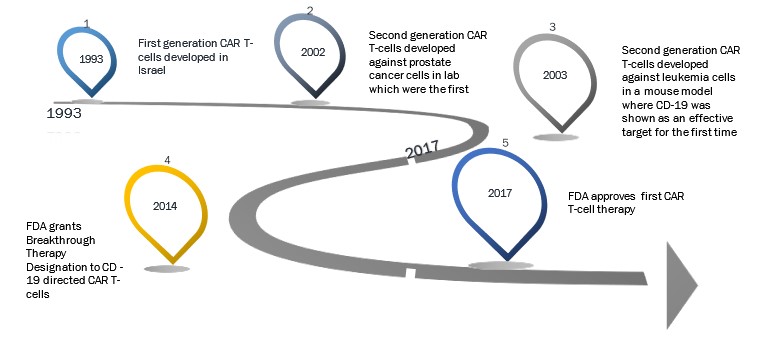
Evolution of CAR T-Cell Therapy[2]
Overview of Regulatory Landscape in the United States for CAR T-Cell Therapies
The Food and Drug Administration (FDA) is responsible for regulating CAR T-cell therapies in the United States. It is important to note the special divisions under FDA that were established to streamline the regulatory processes for the oversight of the development of drugs like CAR T.
FDA’s Departmental Responsibilities
The Office of Tissues and Advanced Therapies (OTAT), a department under the Center for Biologics Evaluations and Research (CBER), is the FDA division primarily responsible for the regulation of CAR T-cell therapies. This includes the review of Chemistry, Manufacturing, and Controls (CMC) data, pharmacology data, and toxicology data. The Office of Compliance and Biologics Quality (OCBQ) is charged with the responsibility of inspection, surveillance, and compliance of a biologic through its lifecycle, which in this case is CAR T-cell therapy. The OCBQ reviews activities such as bioresearch monitoring and establishment inspections. All the statistical and epidemiological evaluation of data is overseen by the Office of Biostatistics and Epidemiology (OBE).[3]
As CAR T is primarily indicated for the treatment of cancers, the Oncologic Drugs Advisory Committee (ODAC) plays a role in the review of the Biologics License Application (BLA). ODAC meetings are designed to be available to the public as an attempt to promote transparency with regards to safety data prior to approval. ODAC reviews the safety and efficacy data and votes for or against the approval of a product based on parameters such as benefit-risk ratio. ODAC’s recommendations are used as guidance and not a directive; hence, FDA is not obligated to follow its recommendation. The Advisory Committee can bring in ad hoc members who are experts specific to the topic of discussion, and these meetings can often serve as a tool of awareness and scientific understanding for healthcare professionals and patients.[4]
FDA can typically place a trial on clinical hold if unexpected or serious safety issues arise. This leads to a trial suspension or delay. During this time, FDA may recommend the sponsor to modify eligibility criteria based on the adverse events. Dosing reevaluation may also be recommended based on the frequency and intensity of the adverse event.[5]
FDA Consultation, Application, and Approval Process
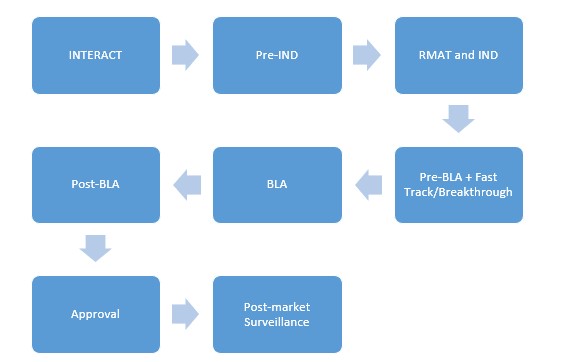
Although CAR T development has dated back to the 1990s, it is still a new arena in the context of first-in-human clinical trials. As we discover serious complications of CAR T therapies in humans and simultaneously learn the benefits that these drugs can bring, developing clinical trial protocols that ensure an efficacious drug dosage and yet safeguard patients from the serious side effects proves to be a challenge. To address such issues, FDA developed a program called INitial Targeted Engagement for Regulatory Advice on CBER producTs (INTERACT) for informal, non-binding advice on the initial stages of innovative investigational product development. This program is designed for a sponsor who has identified an investigational product and has yet to develop an early clinical trial design. A sponsor may, therefore, request an INTERACT meeting early on in the pre-IND (Investigational New Drug) stage as one of the first steps towards achieving regulatory approval.[6]
A pre-IND meeting is highly recommended for the development of CAR T-cell therapy. There is so much we still do not know about CAR T-cell therapy, and a pre-IND meeting can help refine a sponsor’s strategy in identifying the studies required to prove the safety and effectiveness of the product. FDA responds to a pre-IND meeting request within fourteen days and schedules the meeting within sixty days from receipt of request.[7]
The RMAT designation request may be submitted contemporaneously with an IND or as an amendment to an existing IND. FDA is required to respond within sixty days of such a request However, FDA will not grant an RMAT designation if the RMAT criteria are not met, the IND is placed on a clinical hold, or the RMAT request is incomplete.[10]
An IND application must comprise of animal pharmacology and toxicology studies, manufacturing information, clinical protocols, and investigator information. The sponsor is required to wait for thirty days post-IND submission prior to commencing any clinical trial. The sponsor can then plan a pre-BLA meeting with FDA.[11]
A pre-BLA meeting allows the sponsor to dialogue with FDA regarding filing and format issues and address any unresolved concerns to ensure the adequacy of supporting data. An ideal time for a pre-BLA meeting is six months before the targeted BLA submission date. Discussion regarding CMC information is highly recommended. At this time, it is pertinent to discuss risk management and postmarket safety studies. The reason for this meeting is to ensure the sponsor has adequate data to support the approval application and, if not, will then have sufficient time to address inadequacies prior to BLA application. A BLA submission must include form 356h, manufacturing information, applicant information, preclinical studies, clinical studies, and labeling information. In accordance with the Prescription Drug User Fee Agreement (PDUFA), FDA consented to review the BLAs within ten months of the sixty-day filing period. The review period is cut down to six months for priority applications. The PDUFA fee is waived for an orphan designated biologic. Post-BLA submission, FDA reviews the BLA, responds to the sponsor within seventy-four days, and decides if an Advisory Committee meeting is necessary. All the deficiencies identified are addressed during reviews and subsequent meetings.[12]
Expedited Pathways
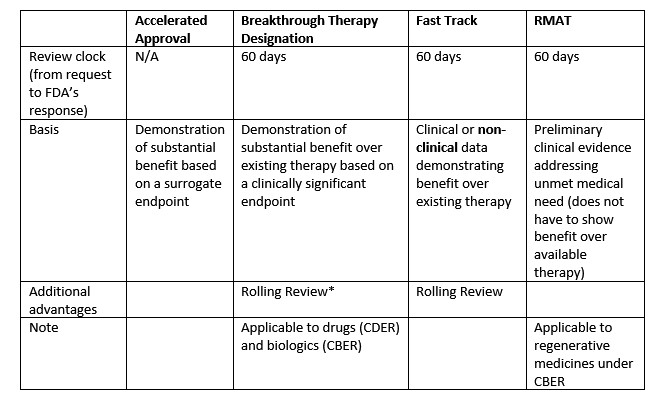
CAR T-cell therapies are targeted to treat potentially fatal diseases with an unmet medical need and hence require a fast turnaround time to gain regulatory approval. There are expedited programs put forth by FDA that enable accelerated development of treatments for life-threatening conditions. Accelerated Approval, Fast Track designation, and Breakthrough designations are explained below.
Accelerated Approval is an expedited process to shorten review time based on a surrogate endpoint. It is essential to understand the difference between surrogate and clinical endpoints to choose an appropriate expedited pathway.
“A surrogate endpoint is a marker, such as a laboratory measurement, radiographic image, physical sign or other measure that is thought to predict clinical benefit but is not itself a measure of clinical benefit. The use of a surrogate endpoint can considerably shorten the time required prior to receiving FDA approval.”[13]
A surrogate endpoint is therefore a predictive measure of clinical benefit rather than a measurable direct clinic benefit itself. In the context of oncology, decrease in tumor size could be considered a surrogate endpoint, while longer survival would be a clinical endpoint. A clinical endpoint can also be derived from proof of effect of a surrogate endpoint.
The Fast Track is another expedited pathway designed to facilitate expedited review of drugs intended to treat a disease or condition that has no existing therapy. If there is an existing therapy, the new drug must show evidence of superiority over the existing therapy in terms of decreasing adverse effects or improving clinical outcome. In this process, the sponsor can have frequent communication with FDA with regards to clinical trial design in addition to the pre-IND meeting and the INTERACT. The idea is to develop an efficient process that brings these drugs to the patient faster but also involves an extensive and integrated review. While the criteria for each of these pathways is similar, there are minor, yet imperative, differences that set them apart. For drugs that can show preliminary evidence on clinically significant endpoints, the Breakthrough Therapy designation is the pathway of choice as opposed to Fast Track where preclinical data is sufficient to fulfill the criteria.
Fast Track and Breakthrough Therapy designations are required to be submitted no later than the time of the pre-BLA meeting. Priority Review can be requested at the time of BLA submission. The timeline of response from FDA is within sixty days of receipt of the request. This timeline is not applicable to accelerated approval pathways.[14]
To summarize, a sponsor would choose the accelerated pathway if data is based on a surrogate endpoint and Breakthrough designation if data is based on preliminary clinical evidence of a clinical endpoint.
Post Approval Requirements
It is mandatory for a sponsor to report any new safety information to FDA when there is adequate data to indicate serious potential risk associated with a drug. FDA may require postmarket studies to assess the risks. The sponsor must submit a study schedule for the postmarket study and periodically report study development.[15]
FDA Approved CAR T-Cell Therapies
Tisagenlecleucel
Tisagenlecleucel is the first FDA-approved CAR T-cell therapy in the United States under the trade name Kymriah, marketed by Novartis Pharmaceuticals Corporation. Tisagenlecleucel is a CD-19 directed autologous immunotherapy indicated for children and young adults up to twenty-five years of age with refractory or relapsed B cell precursor ALL. In adult patients, it is indicated in relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy, high-grade B-cell lymphoma, and diffuse large B-cell lymphoma arising from follicular lymphoma.
Tisagenlecleucel has been awarded orphan drug designation for the treatment of diffuse large B-cell lymphoma, as the indication falls under the category of rare diseases affecting less than 200,000 people in the United States. Pursuant to 21 C.F.R. Part 316, an orphan drug designation was requested by the sponsor and approved by FDA. An orphan drug BLA is exempt from the Pediatric Research Equity Act (PREA). Tisagenlecleucel has also been granted rare pediatric disease designation. Consequently, a priority review has been granted, which involves a shorter review time, i.e., six months. As the drug also qualified for Breakthrough Therapy designation, it was eligible for a rolling review.
An overall remission rate (ORR) was the primary endpoint for a phase II open-label single-arm clinical trial (ELIANA) to determine the safety and efficacy of Tisagenlecleucel in pediatric and young adult patients with R/R B-cell ALL. Minimal residual disease and duration of remission were taken as the secondary efficacy endpoints. Favorable results of 63% complete remission with known complications and acceptable dose led to the approval of Tisagenlecleucel.[16]
FDA has identified long term safety concerns, specifically, the potential for generating replication-competent retrovirus (RCR) and the potential for developing genotoxicity due to limited available data. To address these issues, FDA proposed prospective registry and a long-term follow up study for patients who received Tisagenlecleucel for up to fifteen years post treatment.[17]
Axicabtagene Ciloleucel
Axicabtagene ciloleucel is the second approved gene therapy in the United States, under the trade name Yescarta, marketed by Gilead-Kite Pharma. Axicabtagene ciloleucel is a CD-19 directed autologous immunotherapy indicated for the treatment of relapsed or refractory B-cell lymphoma after two or more lines of systemic therapy, including diffuse large B-cell lymphoma (DLBCL) not otherwise specified, high-grade B-cell lymphoma, primary mediastinal large B-cell lymphoma (PMBCL), and DLBCL arising from follicular lymphoma (FL).
Axicabtagene ciloleucel received orphan drug designation for DLBCL, PMBCL, and FL. It was granted Breakthrough Therapy designation for refractory, aggressive non-Hodgkin’s lymphoma. The approval was based on clinical trial ZUMA-1, where the primary endpoint was a favorable objective response rate (ORR) of 72%.[18]
Similar to Kymriah’s pharmacovigilance proposal, FDA recommended a prospective observational study for fifteen years post-treatment to address long term safety.[19]
Brexucabtagene Autoleucel
On July 24, 2020, FDA approved the third CAR T-cell therapy in the United States, Brexucabtagene autoleucel, under the name Tecartus, marketed by Gilead-Kite Pharma. Brexucabtagene autoleucel is a CD19-directed genetically modified autologous T-cell immunotherapy. It is the first and only CAR T-cell therapy indicated for the treatment of adult patients with relapsed or refractory mantle cell lymphoma (MCL). Tecartus received Breakthrough Therapy designation and Orphan Drug designation. It was approved under the Accelerated Approval Pathway with a Priority Review. The approval was based on results established from ZUMA-2 pivotal trial, where 62% of subjects achieved Complete Remission (CR) after Tecartus treatment.[20]
International Approvals of CAR T- Cell Therapies
European Commission—European Medicines Agency[21]
In 2018, Kymriah and Yescarta were approved by the European Commission (EC) through the PRIority MEdicines (PRIME) program. The PRIME program was designed by the European Medicines Agency (EMA) to build on regulations that support the accelerated assessment of medicines intended to target an unmet medical need. The Committee for Medicinal Products for Human Use (CHMP) and the Committee for Advanced Therapies (CAT) are both responsible for the regulation of CAR T-cell therapies.[22]
EMA mandates post-authorization safety study (PASS) and post authorization efficacy study (PAES). Safety follow up data is to be collected until December 2038. Furthermore, periodic safety update reports (PSUR) are required every six months for zero to two years, annually from two to four years, and lastly, every three years after the four-year mark.[23]
Others
In 2019, Japan’s Ministry of Health, Labor, and Welfare (MHLW) approved Tisagenlecleucel (Kymriah). Similar to FDA’s Breakthrough designation and EU’s PRIME program, Japan introduced SAKIGAKE designation in 2015 for promoting the early application of innovative products.
The timeframe from pre-clinical research to commercialization in market for a regular drug is two months for Phase I/II consultation and twelve months for Phase III review. The time to approval is significantly reduced with SAKIGAKE designation wherein the Phase I/II consultation takes one month, and the Phase III priority review takes six months.[24]
CAR T Infusion Center Prerequisites
Hospitals and healthcare facilities need to meet certain requirements in order to administer CAR T-cell therapy safely. It is highly recommended that such facilities comply with the standards of authorities such as Foundation for the Accreditation of Cellular Therapy (FACT) and Joint Accreditation Committee ISCT-Europe & EBMT (JACIE) and hold formal accreditation.
The medical specialty departments required to oversee the CAR T-cell therapy infusion and its potential adverse effects are hematology and oncology, neurology, emergency medicine, intensive care, immunology, radiology, infectious disease, and pediatrics (if intended to treat pediatric patients). Apart from the specialized physicians, a CAR T infusion team must have a specially trained paramedical team and pharmacist. Typically, a CAR T-cell infusion center must be well equipped to perform apheresis. It is essential to develop a reliable center for CAR T infusion to ensure safety.[25]
Challenges and Complications
Cytokine Release Syndrome (CRS) is one of the most feared and prevalent complications of CAR T-cell therapy. CRS is characterized by immune activation with significantly elevated inflammatory cytokines. There are many grading systems for the severity of CRS. The University of Pennsylvania grading system has four grades, the fourth being life-threatening consequences. Tocilizumab, marketed under the name ActemraTM, is the first FDA approved drug for the treatment of life-threatening CRS. Tocilizumab acts by blocking the IL-6 receptor.[26]
Central Nervous System toxicity can also occur and is termed as CAR T-cell-related encephalopathy syndrome (CRES) or immune effector cell-associated neurotoxicity syndrome (ICANS). This is thought to be due to an ineffective blood-brain barrier in the presence of activated CAR T-cells and neurotoxic substances. On-target off-tumor detection, tumor lysis syndrome, and cytopenia are other adverse effects of CAR T-cell therapy.[27]
Risk Evaluation and Mitigation Strategies (REMS), as mandated by FDA for CAR T, aims to mitigate the CRS and neurological risks by ensuring adequate access to Tocilizumab and meet the infusion center prerequisites as described in the section above.
Future of CAR T-Cell Therapy
Expanding Targets
The current CAR T-cell therapies target biomarker CD-19. Expanding the scope of target molecules can prove to be a potential game changer for CAR T-cell therapy. Lack of CD-19 receptor on cancer cells contributes to relapse. One of the advantages of targeting multiple antigen receptors with a single therapy is to ensure a majority target coverage. At present, in addition to CD-19, CD-22 is being studied to overcome immune escape.[28]
Another target currently under research is the B-cell maturation antigen (BCMA) for myeloma. BCMA is a cell surface receptor that is excessively produced in multiple myeloma. FDA granted orphan drug status to one such BCMA targeted investigational product (CT053 by CARsgen Therapeutics) intended to treat multiple myeloma.[29]
Aiming for CAR T Without CRS
Cytokine Release Syndrome is by far the biggest challenge encountered by CAR T-cell therapy. Yescarta comes with black box labeling that highlights the warning related to the dangers of CRS and other neurological complications. Scientists across the world are working towards a better future of CAR T, which involves lessening the severity of CRS. There are several modifications that are being considered to combat the CRS barrier including dose adjustment, prophylactic measures, and early rise in biomarkers.
Off-the-Shelf/Allogeneic CAR T-Cell Therapy
The currently marketed CAR T therapies are autologous wherein the T lymphocytes are isolated from the patient’s blood. In the near future, allogenic CAR T-cells may be expected to be available for which the storage and transport of the cells will require safety analysis and new stringent regulations. Allogenic CAR T therapy is being explored to overcome the limitations associated with autologous therapy. The limitations are related to the laborious manufacturing time and effort for developing personalized CAR T treatments that are specific to that particular patient. As a result of personalized manufacturing, the prices of these therapies are astronomically high. As with any innovative process, allogenic therapy does not come without potential dangers. Allogeneic CAR T-cells are derived from a donor and thus require compatibility testing with the patient, similar to the organ transplant process. In August 2018, FDA accepted the first non-gene edited allogeneic CAR T IND application for a candidate CYAD-101 studied by a biopharmaceutical company, Celyad.24
Alternative to Chemotherapy and Stem Cell Transplant
CAR T-cell therapy is shifting the paradigm of oncology treatment. CAR T is traditionally given after chemotherapy, but that may change soon with the rapid advancement in research.
Use in Solid Tumors
CAR T-cell therapy has shown promising results in the treatment of hematological malignancies. The expansion of its indications to solid tumors is making progress in clinical trials worldwide.[30] An investigational CAR Tin the news for solid tumors is a CAR T investigated by Memorial Sloan Kettering researchers with promising Phase I results for the treatment of mesothelioma.[31]
Conclusion
It is important to note that CAR T therapy is indicated not only in adults but also in pediatric patients and young adults, which raises long term safety concerns such as effects of these drugs on growth, development, and fertility. Additionally, potential for secondary malignancies remains a concern in all age groups. Limited data due to fast-paced trials prevents us from definitively concluding if the aforementioned risks are minimal. The pharmacovigilance plan currently in effect for these therapies is a step in the right direction. However, it could be beneficial to consider postmarket efficacy studies similar to EMA to accurately assess the benefit-risk ratio of these therapies. Furthermore, mandating Phase 0/early-Phase I studies as we see more scientific advancements will help exclude certain high-risk populations while exposing fewer subjects.
Appendix
Supplemental Data
Source: https://clinicaltrials.gov/ct2/results/map?term=CAR+T&recrs=a&phase=012345&map=
[1] Summary Basis for Regulatory Action – KYMRIAH, U.S. Food and Drug Administration 1 (Aug. 30, 2017), https://www.fda.gov/media/107962/download. (Accessed August 4, 2020).
[2] CAR T Cells: Timeline of Progress, Memorial Sloan Kettering Cancer Center (2020), https://www.mskcc.org/timeline/car-t-timeline-progress. (Accessed July 7, 2020).
[3] Op. Cit. 1.
[4] Ariadna Tibau, Alberto Ocana, Geòrgia Anguera, et al., Oncologic Drugs Advisory Committee Recommendations and Approval of Cancer Drugs by the US Food and Drug Administration, 2 JAMA Oncol. 744 (2016), https://jamanetwork.com/journals/jamaoncology/fullarticle/2497879.
[5] IND Application Procedures: Clinical Hold, U.S. Food and Drug Administration (2020), https://www.fda.gov/drugs/investigational-new-drug-ind-application/ind-application-procedures-clinical hold#:~:text=A%20clinical%20hold%20is%20an%20order%20issued%20by,IND%20application%20may%20be%20placed%20on%20clinical%20hold. (Accessed Nov. 9, 2020).
[6] INTERACT Meetings, U.S. Food and Drug Administration (2020), https://www.fda.gov/vaccines-blood-biologics/industry-biologics/interact-meetings-initial-targeted-engagement-regulatory-advice-cber-products. (Accessed July 3, 2020).
[7] Small Business and Industry Assistance: Frequently Asked Questions on the Pre-Investigational New Drug (IND) Meeting, U.S. Food and Drug Administration (2020), https://www.fda.gov/drugs/cder-small-business-industry-assistance-sbia/small-business-and-industry-assistance-frequently-asked-questions-pre-investigational-new-drug-ind. (Accessed July 3, 2020).
[8] 21 U.S. Code § 356 (2020).
[9] Expedited Programs for Regenerative Medicine Therapies for Serious Conditions Guidance for Industry, U.S. Food and Drug Administration Center for Biologics Evaluation and Research 5–6 (Feb. 2019), https://www.fda.gov/media/120267/download.
[10] Id. at 8.
[11] IND Application Procedures: Overview, U.S. Food and Drug Administration Center for Drug Evaluation and Research (Oct. 9, 2015), https://www.fda.gov/drugs/investigational-new-drug-ind-application/ind-application-procedures-overview. (Accessed August 17, 2020).
[12] CDER 21st Century Review Process: Desk Reference Guide, U.S. Food and Drug Administration 6–10 https://www.fda.gov/media/78941/download. (Accessed August 10, 2020).
[13] Accelerated Approval Program, U.S. Food and Drug Administration (Oct. 26, 2020), https://www.fda.gov/drugs/information-health-care-professionals-drugs/accelerated-approval-program.
[14] Expedited Programs for Regenerative Medicine Therapies for Serious Conditions Guidance for Industry, U.S. Food and Drug Administration Center for Biologics Evaluation and Research 4, 9 (Feb. 2019), https://www.fda.gov/media/120267/download. (Accessed Nov. 9, 2020).
[15] Notice to Industry: Postmarketing Requirements – Postmarket Studies and Clinical Trials, U.S. Food and Drug Administration (Feb. 2, 2016), https://www.fda.gov/drugs/guidance-compliance-regulatory-information/notice-industry-postmarketing-requirements-postmarket-studies-and-clinical-trials. (Accessed Nov. 8, 2020).
[16] Summary Basis for Regulatory Action, supra note 1, at 1, 7.
[17] Id.
[18] BLA Clinical Review Memorandum Yescarta, U.S. Food and Drug Administration 3, 47, https://www.fda.gov/media/109149/download. (Accessed Aug. 10, 2020).
[19] Id.
[20] FDA Approves First Cell-Based Gene Therapy for Adult Patients with Relapsed or Refractory MCL, U.S. Food and Drug Administration (July 24, 2020), https://www.fda.gov/news-events/press-announcements/fda-approves-first-cell-based-gene-therapy-adult-patients-relapsed-or-refractory-mcl. (Accessed July 28, 2020).
[21] Applicable Regulations: Commission Directive 2003/63/EC amending Directive 2001/83/EC of the European Parliament and the Council on the Community code relating to medicinal products for human use.
[22] First Two CAR-T Cell Medicines Recommended for Approval in the European Union, European Medicines Agency (June 29, 2018), https://www.ema.europa.eu/en/documents/press-release/first-two-car-t-cell-medicines-recommended-approval-european-union_en.pdf. (Accessed July 3, 2020).
[23] Id.
[24] Strategy of SAKIGAKE, Japan Ministry of Health, Labour, and Welfare, https://www.mhlw.go.jp/english/policy/health-medical/pharmaceuticals/140729-01.html. (Accessed July 3, 2020).
[25] Christine Chomienne, Jorge Sierra, Hermann Einsele & Ulrich Jäger, EHA Guidance Document: The Process of CAR-T Cell Therapy in Europe, 3 HemaSphere e280 (Aug. 2019), http://dx.doi.org/10.1097/HS9.0000000000000280.
[26] Drug Approval Package: ACTEMRA (tocilizumab), U.S. Food and Drug Administration (Apr. 13, 2018), https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/125276Orig1s114TOC.cfm. (Accessed July 3, 2020).
[27] Lucrecia Yáñez, Miriam Sánchez-Escamilla & Miguel-Angel Perales, CAR-T Cell Toxicity: Current Management and Future Directions, 3 HemaSphere e186 (Apr. 2019), https://journals.lww.com/hemasphere/Fulltext/2019/04000/CAR_T_Cell_Toxicity__Current_Management_and_Future.5.aspx. (Accessed July 3, 2020).
[28] Bianca Santomasso, Carlos Bachier, Jason Westin, Katayoun Rezvani & Elizabeth Shpall, The Other Side of CAR T-Cell Therapy: Cytokine Release Syndrome, Neurologic Toxicity, and Financial Burden, 39 American Society of Clinical Oncology Educational Book 433 (May 17, 2019), https://ascopubs.org/doi/10.1200/EDBK_238691. (Accessed July 3, 2020).
[29] CARsgen Announces Investigational CAR-T Therapy CT053 Granted RMAT Designation by the U.S. FDA for R/R Multiple Myeloma, BioSpace (Oct. 28, 2019), https://www.biospace.com/article/releases/carsgen-announces-investigational-car-t-therapy-ct053-granted-rmat-designation-by-the-u-s-fda-for-r-r-multiple-myeloma/. (Accessed July 3, 2020).
[30] Flora Southey, US FDA Approves First Non-Gene Edited Allogeneic CAR T-Cell Candidate for Trials, Biopharma (Aug. 15, 2018), https://www.biopharma-reporter.com/Article/2018/08/15/US-FDA-approves-first-non-gene-edited-allogeneic-CAR-T-cell-candidate-for-trials. (Accessed July 3, 2020).
[31] Diane Seimetz, Karl Heller & Jan Richter, Approval of First CAR-Ts: Have we Solved all Hurdles for ATMPs?, Cell Medicine (Jan. 22, 2019), https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6343443/.
 SHRADDHA KOMANDURI is a regulatory specialist II at the Penn State Cancer Institute working in the Hematology/Oncology Clinical Trials Department, where she previously served as a clinical research associate. She is a medical graduate from India with a Master of Science degree in regulatory science from Johns Hopkins University. She would like to extend her thanks to Dr. Thomas Colonna and Doris Shank for their invaluable guidance throughout the process.
SHRADDHA KOMANDURI is a regulatory specialist II at the Penn State Cancer Institute working in the Hematology/Oncology Clinical Trials Department, where she previously served as a clinical research associate. She is a medical graduate from India with a Master of Science degree in regulatory science from Johns Hopkins University. She would like to extend her thanks to Dr. Thomas Colonna and Doris Shank for their invaluable guidance throughout the process.Update Magazine
Winter 2020