Focusing on the Patient: Implementation of Key 21st Century Cures Provisions and Recommendations for the Future
By Nick Manetto, Lauren Bloch, Annie Kennedy, and Tim Franson
Overview
Over the past decade, Congress has enacted several laws to infuse the voice and perspective of the patient into the therapy development and regulatory review process. Known as patient engagement or patient-focused drug development, these laws and their related Food and Drug Administration (FDA) regulations, guidance documents, and other regulatory activities have had a marked impact in informing agency actions, including by those tasked with reviewing and approving product applications.
The arc of patient engagement stretches back to the FDA Safety and Innovation Act (FDASIA, PL 112-144), which was enacted in 2012, and to metrics contained in the corresponding Prescription Drug User Fee Act (PDUFA V) agreement.[1] This arc has subsequently extended into the 21st Century Cures Act (PL 114-255),[2] enacted in late 2016, and on into the FDA Reauthorization Act (PL 115-52), enacted in August 2017.[3] As we write, interest in patient engagement policy continues to expand as legislators, regulators, industry, and patient stakeholders turn their attention to PDUFA VII, which must be enacted by September 2022.
While patient engagement is a broad, growing category, this article will review two provisions of the 21st Century Cures Act: Sections 3001 (Patient Experience Data) and 3002 (Patient-Focused Drug Development Guidance), which sought to achieve two fundamental goals regarding patient engagement policies:
- Creating more transparency as to how patient information is being considered as part of medical product review processes; and
- Providing clearer direction and guidance to stakeholders interested in developing patient engagement tools, including direction and guidance on processes and standards for producing instruments appropriate for regulatory review.
Combined, these two provisions sought to advance the field of patient engagement by providing greater clarity as to how patient experience data and related information are used by FDA review staff and to clarify the agency’s standards for developing qualified regulatory tools and FDA’s process for doing so. Given the time and economic resources associated with developing scientifically rigorous patient engagement tools, answers to both questions were seen as necessary to further nurture and develop the field of patient engagement.
As we near the four-year anniversary of the enactment of 21st Century Cures and the recent launch of the process to craft the next user fee agreement (PDUFA VII), this article examines the legislative history behind Sections 3001 and 3002 and how these important provisions have been implemented by FDA. It also analyzes the strengths as well as the deficiencies associated with both provisions and, informed by this analysis, offers recommendations for policies and practices to further strengthen the implementation of these provisions and for the enactment of additional changes through PDUFA VII or other related legislation.
Legislative History & Intent—PFIA and Sections 3001 and 3002
Following the enactment of FDASIA in 2012, a number of patient advocacy organizations sought to leverage new FDASIA authorities to advance the field of patient engagement. One of the most proactive communities was the Duchenne Muscular Dystrophy (DMD or Duchenne) community. During a late-2013 patient forum attended by multiple FDA officials, the issue of a potential patient-led process to develop a draft guidance was broached.
At that time, FDA had not approved any therapies for Duchenne, though some candidate therapies were moving toward later-stage clinical evaluation (note: generally, this article uses the terms drugs and therapies interchangeably). The community saw several opportunities in the space, including a need for greater guidance and clarity from FDA to industry for the development of Duchenne therapies. About one year after FDASIA was enacted, the advocacy organization Parent Project Muscular Dystrophy (PPMD) initiated a comprehensive and multi-stakeholder effort to develop a draft guidance that would be submitted to FDA in the hopes of spurring the agency to issue such a guidance document of its own.[4]
The effort to develop such a comprehensive document played out over more than a year and culminated with PPMD submitting the draft guidance to FDA in June 2014.[5] This action spurred FDA to issue draft guidance a year later,[6] and FDA issued the final guidance in February 2018.[7] PPMD’s work led to high levels of visibility and accolades for the organization, particularly as the legislative effort to develop the 21st Century Cures Act gained traction during the summer of 2014. While the attention was positive, a fundamental question for PPMD and others was how any patient engagement content—meetings, meeting reports, patient-preference studies, and guidance documents—would actually impact FDA’s product reviews.
With this in mind, PPMD began to have conversations with several congressional offices to explore what policy solutions may exist to answer the fundamental question of how patient engagement data was informing regulatory reviews, particularly in the review of specific applications. In June 2015, as Duchenne families gathered in Washington, DC for the annual PPMD Connect Conference, Senators Roger Wicker (R-MS) and Amy Klobuchar (D-MN) introduced S. 1597, the Patient-Focused Impact Assessment (PFIA) Act. The PFIA Act would become Sections 3001 and 3002 of the 21st Century Cures Act.[8] The PFIA Act enjoyed bipartisan support from its introduction, with a bipartisan group of four additional original cosponsors.
The bill as introduced contained two main sections:
1) A requirement for FDA to publish a publicly available summary about how patient experience information, if any, was factored into a product’s review; and
2) A requirement for FDA to issue guidance to inform development of patient engagement tools.
The overall objective of the legislation was to require some level of transparency and accountability as to whether and how such patient experience information is used in the review process while also requiring clear guidance from FDA to inform those interested in developing such tools as to how they should go about the work. Importantly, the PFIA Act never sought to mandate that FDA use any patient engagement data. Additionally, it focused on requiring that such information be specified as part of the drug data package FDA issues upon approval of a newly approved drug but did not address how such information may be used by FDA to decide not to approve an application and to issue a complete response letter.
As the PFIA Act advanced through the legislative process, including clearing the Senate Health, Education, Labor, and Pensions (HELP) Committee in April 2016 and eventually being included in the 21st Century Cures Act, it underwent some amendments.[9] Notably, these amendments included a shift to the term “patient experience data” (PED) and a more global approach to defining content that would fall under the moniker of patient experience data. As 2016 moved forward and work on the 21st Century Cures Act advanced, the PFIA Act was incorporated into Sections 3001 and 3002 of the final 21st Century Cures Act. As noted above, Section 3001 focused on the reporting component, referred to in the final law as a statement of patient experience, and Section 3002 directed FDA to issue guidance documents to inform development of patient experience data and tools.[10]
Implementation and Evolution of the Patient Experience Table—2017 to 2020
Section 3001(a) of the 21st Century Cures Act requires FDA, following its approval of a drug, to “make public a brief statement regarding the patient experience data and related information, if any,” submitted to, and reviewed by, FDA.[11] It also included the term “at least 180 days after the date of enactment,” meaning that tables would not be required under the law until mid-June 2017, a reason why our review began with 2018 approvals. Given the late-2016 enactment of the 21st Century Cures Act, FDA spent much of 2017 working to implement the Act’s provisions, including Sections 3001 and 3002. The “brief statement” eventually took the form of a table, known as the patient experience data table, that allowed FDA reviewers to indicate the types of PED they reviewed for the new drug application.
Figure 1: Patient Experience Data Table from Multi-Disciplinary Review of Hemlibra, Obtained from FDA Website
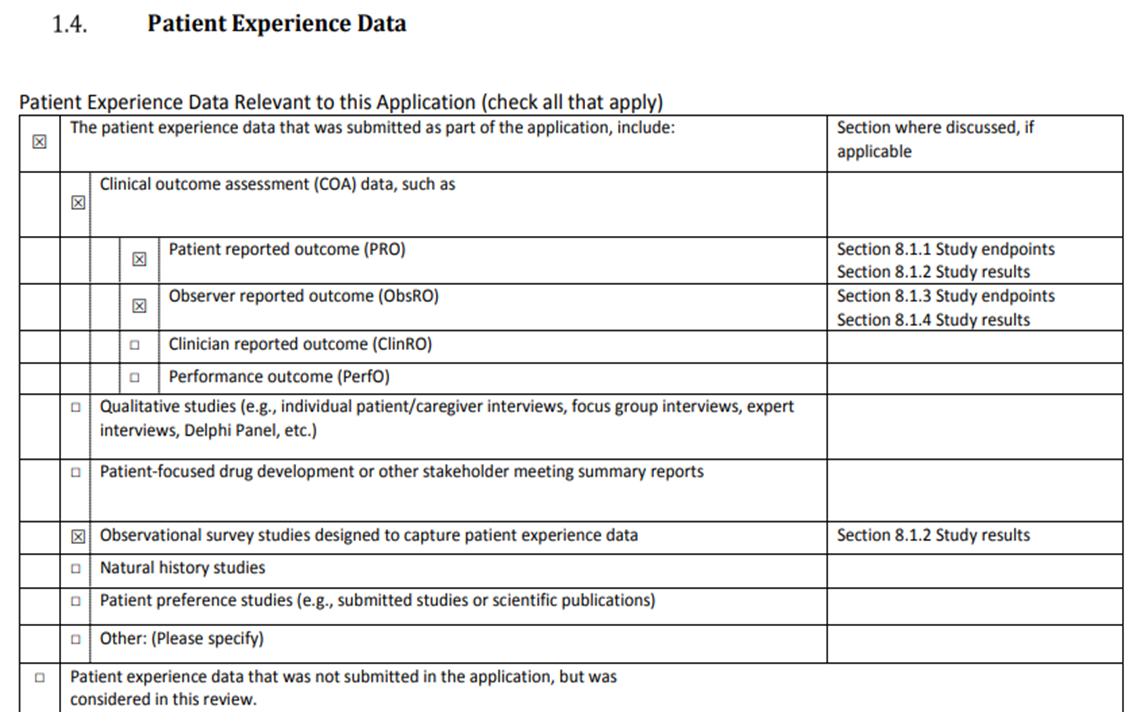
The first drug approval package containing the patient experience data table was for Hemlibra, a Genentech drug approved in November 2017 to treat bleeding in adults and children who have hemophilia A.[12]By statute and its 180-day (six month) implementation timeframe, the information was required to be provided by FDA following its approvals of applications submitted on May 13, 2017, or later. By late 2017, review packages for approved therapies began to contain the earliest published versions of the patient experience data table and to reference the table within the table of contents.
Inclusion of Patient Experience Data—2018 through September 2020
The PED table represents FDA’s effort to meet the intent of the 21st Century Cures Act’s requirement that the “brief statement” must address “patient experience data; information on patient-focused drug development tools; and other relevant information, as determined by the Secretary.”
Based on our review of drug data packages for novel drug approvals from January 2018 through September 2020 (N=147), 62% of review packages for approved new therapies met this requirement.[13] The review was done by searching each data package for inclusion of a statement of patient experience.
Of the packages that contained a patient experience section and the PED table, 73% included tables indicating that at least one type of PED was used in the review of the overall application. In the remaining cases, reviewers either checked the box indicating that PED was not considered as part of the application or left the table blank completely, presumably indicating that no such data were included within the application.
Not surprisingly, many of the reviews that did not consider PED were from early 2018. The authors of this article understand the long-term nature of drug development and the reality that applications reviewed and approved during this window were advanced long before Section 3001 was enacted and, as mentioned previously, note the 180-day implementation threshold. Unfortunately for those seeking to see more robust use of patient experience information in regulatory decision-making, the trajectory has not been entirely upward.
Based upon our review and as shown in Figure 2, 23% of data packages associated with 2019 approvals and 48% of packages associated with drugs approved during the first nine months of 2020 did not include PED information. While the numbers could change if/as more data are posted, the overall trends suggest there is still work to further efforts to include relevant PED within applications and to ensure review of such by agency reviewers.
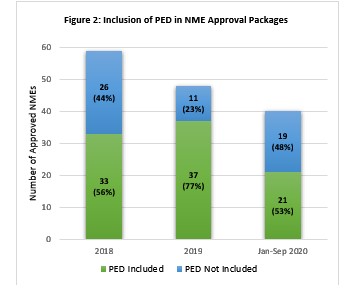
Patient Experience Data Submitted
Based on our review, the vast majority of PED used in reviews was submitted with the application (88%) rather than coming from separate sources considered by FDA (12%). As shown in Figure 3, clinical outcome assessment (COA) data comprised the large majority (81%) of PED submitted with applications. Patient-reported outcomes (PROs) were the most commonly identified subset of COA.
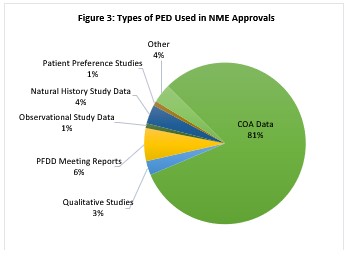
Of the reviews for which FDA considered separate sources of PED not submitted with the application, only six considered reports from patient-focused drug development (PFDD) or other stakeholder meetings, despite the fact that 23% of drugs approved were indicated for conditions for which an FDA-led or externally-led PFDD meeting was held. Other sources of data referenced by FDA included an Advisory Committee meeting, an agency meeting with a patient advocacy organization, and literature reviews.
Many PED tables include references to the name or number of specific sections of the review that included the data. However, in our subsequent review of the sections that were cited, we found significant variation as to how such data were identified, if at all, in the review sections. For example, while many PED tables indicated patient experience data was included within clinical outcomes data, including patient-reported outcomes, or that patient engagement meetings or meeting reports informed the approval package’s section on the overview of the condition and current treatments, most of the reviews fell short of explicitly referencing the patient engagement data in the descriptive sections. Upon subsequent review of the descriptive sections referenced, it was at times possible to find indicators of PED, such as identification of a patient-reported outcome or agency discussion on inclusion of such information. But in other cases, particularly in the summary of the condition and currently available therapies section, the use of patient engagement data was not clearly evident.
A few of the reviews stood out for the level of detail used by FDA to describe the patient engagement data it reviewed as part of the application. For example, the approval package for the drug Ongentys, approved in late April 2020 to treat Parkinson’s Disease, included within the clinical outcomes section references to patient diaries that were used to track secondary outcomes. Additionally, the approval package’s analysis of the condition included multiple references to the September 2015 FDA-convened PFDD meeting on Parkinson’s along with data from the corresponding meeting report.[14]
Similarly, the analysis of condition section of the approval package for Takhzyro, a drug approved in August 2018 to treat hereditary angioedema, described the overarching themes from the PFDD meeting convened by FDA in September 2017.[15] And the October 2019 approval package for the cystic fibrosis drug Trikafta is one of the few we found that explicitly referenced the work done by a patient advocacy organization. The approval package cited data, within the analysis of condition section, from the Cystic Fibrosis Foundation’s patient registry.[16]
Some other reports cited submitted patient-reported outcomes data, while going on to state that the agency viewed such data as being exploratory because of various shortcomings. For example, the August data associated with approval of the drug BLENREP to treat multiple myeloma included a discussion about patient-reported outcomes that the applicant summarized as reflecting worsening of eye symptoms and maintenance of overall symptoms.[17] FDA, however, asserted in the document that the quality of life data was limited for several reasons and that the agency disagreed with the interpretation of the sponsor and stated that the trial “was neither designed nor powered to assess maintenance of QoL.” While some may view examples like this as being negative, we take a more positive outlook and consider feedback like that as being consistent with the intent of the statute, particularly to provide greater guidance to those developing or considering developing PED. Such feedback is important because it makes clear the rigor FDA expects within any PED data and provides guidance to interested stakeholders.
Section 3002 and FDA Progress on Guidance Development
To drive the use of patient experience data in drug development, Section 3002 of the Cures Act requires FDA to develop guidance documents on how these data could be collected and used. After first developing a plan to publish a series of six guidance documents by the end of FY 2021, FDA has begun to follow a series of steps for each: holding a public workshop to collect initial input, publishing a draft guidance, collecting comments on the draft, and publishing a final guidance. To date, FDA has completed this process for one of the six guidance documents. The agency has also published two draft guidances and has held public workshops to support the development of the three remaining guidance documents (see Addendum 1). Five of the six guidances primarily focus on front-end processes for using patient data in drug development and regulatory decision-making, namely guidance on how to identify, collect, and submit these data.
The sixth guidance document required by Section 3002 focuses on how FDA will use PED submitted as part of applications and, per FDA’s original plan, was slated for draft publication in the second quarter of 2020. Given its focus on agency use, it is perhaps the most important guidance document in the series and will provide additional context for therapy sponsors and patient advocacy organizations seeking to collect patient information that is useful to FDA in its reviews. FDA has stated that this guidance will also touch on how patient experience data factor into reviewers’ benefit-risk assessments. In May 2019, FDA, in partnership with the Duke Margolis Center for Health Policy, convened a public workshop to facilitate the development of this guidance document. Notably, of the more than twenty panelists who spoke at the meeting, only one was from a patient advocacy organization.
Findings and Analysis
Overall, FDA has made significant progress in implementing Sections 3001 and 3002 of the 21st Century Cures Act. At the time of this analysis, about three-and-a-half years have passed since the law was enacted in December 2016. This progress has occurred despite having a presidential transition, three commissioners, and one acting commissioner during this time. The progress has also occurred while the agency has found itself on the frontline of the nation’s ongoing response to the novel coronavirus (COVID-19) pandemic. Based on our analysis, we offer the following takeaways on implementation of Sections 3001 and 3002. These will be followed by some thoughts as to recommendations to further strengthen both policies, which could include actions by FDA as well as by Congress.
Section 3001: Patient Experience Data
FDA has followed the intent of the statute in developing a succinct checklist that summarizes the patient engagement data reviewed as part of a drug application. The table captures key PED categories and allows for reviewers to also note where such content is found in the review itself. It is fairly user-friendly and something that a patient, consumer, or non-regulatory professional could review and digest.
Although the PED table provides a good overview of the types of PED that were submitted with or considered in the review of an application, the completeness and quality of how the table is utilized by FDA varies significantly. We note variation in how reviewers identify and quantify patient engagement data, particularly instances in which data are noted in the summary table yet not clearly delineated in the sections cited. The most robust approval packages include discussion of the PED in the report sections, such as how the data informed the review and whether FDA found the data of limited utility.
There are also other variations with respect to the PED table, including the location of the table within the approval package and when the information becomes publicly available. For example, our review found the PED table, when included, was typically located within the clinical or multi-disciplinary review documents, although not always in the same section of those documents. The review packages for some more recently approved drugs were posted a month or more after the approval letter and label were posted, even though complete information was in some cases available for drugs approved more recently. This raises questions as to when review packages must be posted and if there is often a lag in the full package becoming publicly available. The seeming lack of standardization impedes transparency around the use of PED and therapy approvals, in general.
Although there are still opportunities to improve the quality and use of the PED table, FDA has made improvements in response to recommendations from stakeholders. These improvements have included having sponsors and other stakeholders pre-populate the table and cite references to patient experience data sources within the submission, and FDA having a standard placement of the summary table within the approval package.
Section 3002: Patient-Focused Drug Development Guidance
FDA has made relatively rapid progress in developing these guidance documents and has provided helpful information on the types of patient information that can be submitted with therapy applications and the methods for collecting that information. Despite this progress, it seems unlikely that the agency will complete the full guidance series by the end of FY 2021. This is problematic not only because of the delay, but also because one of the remaining guidance documents focuses on how FDA will use PED. Without clear direction from the agency on this important issue, it remains difficult for therapy developers and patient organizations to fully understand how the provision of patient experience data factors into FDA’s assessment of benefits and risks. This in turn may lead stakeholders to reconsider whether or not to commit the resources necessary to develop scientifically rigorous patient engagement data, chilling advancement of the field.
Conclusion and Recommendations
This analysis was conducted as stakeholders, including FDA, were beginning the multi-year process to develop the next user fee package, due by September 2022, and as members of Congress are considering a second iteration of the 21st Century Cures Act. With the PDUFA VII process kicked off and the anticipated legislative focus on it that will come during the upcoming 117th Congress, we offer the following recommendations informed by our analysis. These include actions FDA can take within current authorities as well as authorities Congress would need to enact.
Agency Actions
Consistent with our findings above, FDA should commit to cross-center and cross-division internal education and capacity building to standardize how the PED summary tables are completed and how PED is referenced within the larger approval packages. Specific actions could include:
- Standardizing the process for filling out the PED table, particularly when noting inclusion of PED data.
- Requiring explicit references to PED data, when noted in the table, within the relevant sections of the review.
- Ensuring the PED table is included in the same document within every data package and consider including it as a standalone document for ease of finding.
- Committing to include more explanatory text when referencing PED data, particularly in explaining how the data factored into the review process or why the agency did not accept data or considered it exploratory in nature, to provide valuable feedback to stakeholders.
- Providing additional guidance as to when patient experience data may be submitted by sources independent of the sponsor and the types of data that may be submitted.
Additionally, FDA should commit to working with stakeholders to solicit feedback as to the current structure and contents of the table to regularly update the table going forward. This could be done in part through the process of developing the sixth PFDD guidance about benefit-risk assessment throughout the medical product lifecycle. The table would ideally reflect the evolution of PED and include the most relevant categories of data being submitted. This engagement should include a particular focus on sponsors to ensure they are educated about Section 3001 requirements and how to present or highlight PED contained in their applications.
To help increase transparency and awareness of how PED has been included within regulatory reviews, FDA should consider publishing an annual summary. Rather than create a new document, this information would seemingly fit within the agency’s annual report of new drug approvals.[18] Pertinent information would include information like what this article is seeking to capture and could be pulled from the PED tables thus placing little to no additional reporting burdens on the agency.
Although the analysis for this article focused on products under the purview of the Center for Drug Evaluation and Research (CDER) given the statutory language of Section 3001, the standardized processes for using the PED summary table should apply to reviews of products by the Center for Biologics Evaluation and Research (CBER) and Center for Devices and Radiological Health (CDRH), as well. Ideally, FDA would focus on driving consistent use of PED across divisions and centers while being careful to not create any unnecessary burdens at the review level.
Ultimately, FDA review divisions are responsible for reviewing applications for new therapies and determining whether the submitted data effectively demonstrate the safety and efficacy required for that therapy to be approved. In order for the patient voice to be truly represented in regulatory decision-making, staff at the review divisions must have access to and understand the value of PED and related information. Although some review divisions have been very actively involved in patient engagement, this seems to be driven largely by the work of individual FDA staff members rather than coordinated efforts on the part of the divisions. FDA should consider ways to better ensure review teams across FDA centers are aware of, have ready access to, and are using PED in their reviews of candidate therapies.
As FDA considers these and other actions to improve the use of PED in drug application reviews, the agency should also consider how best to foster the collection and use of PED outside of sponsor submissions. Currently, FDA permits stakeholders to submit resources related to patient experience data such as proposed draft guidance documents, natural history studies, and reports from externally led patient-focused drug development meetings. FDA does not endorse or vouch for the quality of these resources, but makes them available on a dedicated page[19] on FDA’s website. This page is somewhat difficult to find, however, and at the time of this writing, it appears it is not updated frequently. There is also no indication of whether the externally developed resources are shared with key FDA staff, such as reviewers, who may derive the greatest benefit by having access to additional information on the experiences of people living with various diseases and conditions. To better emphasize the importance of using PED throughout the drug development lifecycle, FDA should consider ways to improve its processes for collecting PED for purposes other than the review of a candidate therapy.
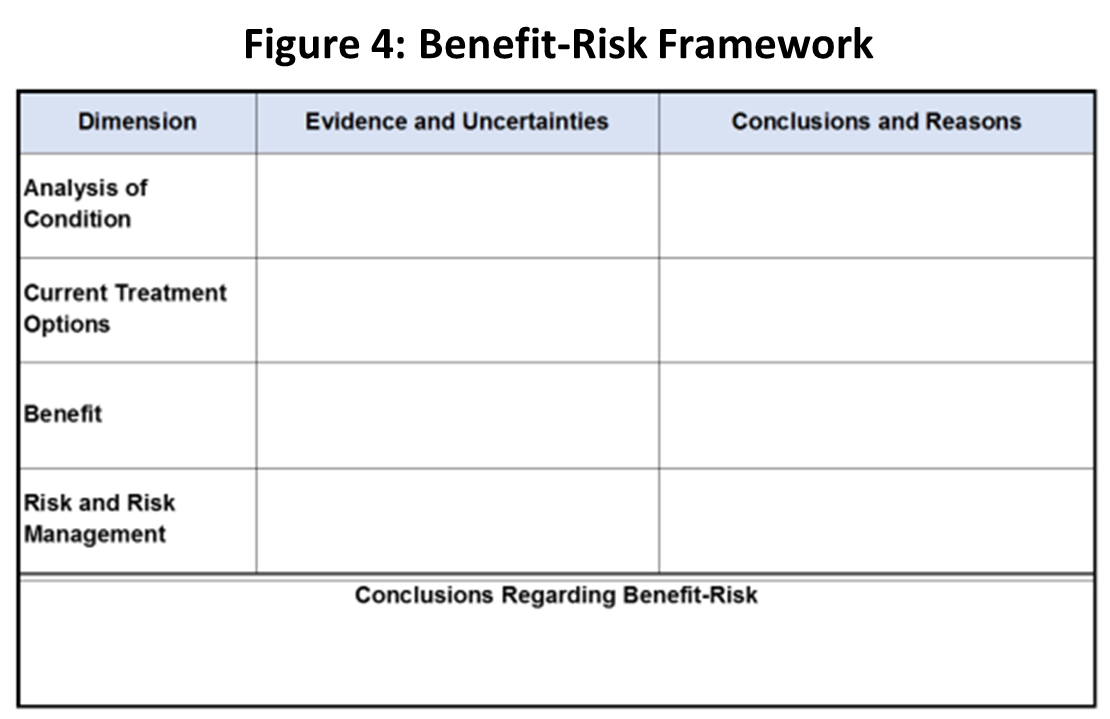
Legislative Actions
FDA should be applauded for its acceptance of patient engagement and its efforts to incorporate patient experience data into reviews, but we note this information is not currently required to be part of the agency’s benefit-risk framework (Figure 4). This framework represents the core of FDA’s decision-making processes and summarizes the key factors that contribute to therapy approvals. With the sixth PFDD guidance document, FDA should provide additional information on how PED factors into the benefit-risk assessment, though the agency has not given any indication that the framework will be modified. Bipartisan legislation known as the BENEFIT Act (S. 3385)[20] would address this gap by including PFDD and related data within the framework. Doing so would serve as a signal to both FDA and medical product sponsors that approvals hinge, in part, on information from patients who represent the population that would ultimately use those products.
To drive consistency in the use of PED across FDA centers, Congress should consider expanding Section 3001 to require a similar summary about patient experience data within review packages of newly approved medical devices. The authors note that CDRH has long been a leader in the field of patient engagement, including by establishing in 2016 a patient advisory committee to advise the center.[21] Notably, when CDRH approved Maestro, a medical device used to achieve weight loss, this approval was often noted as being the first in which a patient preference study factored into an agency review and approval.[22] Additionally, as the pipeline for gene therapies continues to grow, there is also increasing interest in how CBER can incorporate patient information into its review processes.
Requiring the inclusion of a PED table in all medical products and devices approved by FDA would help to drive increased recognition of the value of patient information across the entire agency.
Additionally, Congress should consider building upon existing law to require that patient experience information be incorporated into product labeling, particularly patient inserts. Although inclusion of these inserts is voluntary for labeling of most products, adding a statement about patient experience data would carry forward the goal of making patient information more accessible. Doing so could also aid prescribing physicians and patient users who may find the perspective and experiences of other patients useful in informing their own decision making.
Congress should also consider ways to obtain information on patient experience data from applications that receive complete response letters. The authors recognize the challenges with confidential data pertaining to non-approved applications and that careful consideration would need to be given to any potential solutions. However, we recognize that the data from approved drugs provides only a partial picture into how PED is used as part of the regulatory review process and that information from applications that receive complete response letters may be informative to stakeholders. Ultimately, such data would round out and present a more complete picture, and we believe the policy idea merits consideration, such as using de-identified or composite observations.
Finally, while not connected directly to Sections 3001 and 3002 of the 21 Century Cures Act, we urge lawmakers to move toward creating a bridge from using patient engagement in the FDA regulatory space to using this same type of perspective to inform payer coverage and access policies, particularly for Medicare, Medicaid, and other public payers. Earlier this year, in their concept paper for 21st Century Cures 2.0, Congressman Fred Upton and Congresswoman Diana DeGette included sections to improve FDA-CMS communications and modernize CMS. This is an encouraging start that could and should be nurtured to include policies specific to patient experience and patient engagement.
Conclusion
Based on our analysis, progress has been made in implementing Sections 3001 and 3002 of the 21st Century Cures Act. The statement of patient experience first appeared in the review package of an approved product within one year of enactment of the law, and FDA has issued a number of guidances required by Section 3002. At the same time, opportunities exist to further strengthen these policies through both agency actions and future legislation. Such actions will help further refine and advance the field of patient engagement toward a vibrant ecosystem in which stakeholders develop more rigorous data and tools and in which the agency factors such information into its decision-making in even more meaningful ways. As policymakers consider proposals for the next round of user fee negotiations and related legislation, we hope consideration will be given to the recommendations we have put forward here.
Addendum 1: Status of PFDD Guidance Series
The table below lists the guidance documents FDA is developing in response to the requirements in Section 3002(c) of the 21st Century Cures Act. The dates in the Original Timeline column refer to those announced in the FDA’s Plan for Issuance of Patient-Focused Drug Development Guidance, published in May 2017.
|
Guidance Title |
Original Timeline |
Status |
|
PFDD Guidance 1: Collecting Comprehensive and Representative Input |
Final guidance: Q1 2020 |
Final guidance: June 2020 |
|
PFDD Guidance 2: Methods to Identify What is Important to Patients |
Draft guidance: Q2 2019 Final guidance: Q1 2021 |
Draft guidance: September 2019 |
|
PFDD Guidance 3: Select, Develop, or Modify Fit-for-Purpose Clinical Outcome Assessments |
Public workshop: Q4 2019 Draft guidance: Q2 2020 Final guidance: Q4 2021 |
Public workshop: October 2018 |
|
PFDD Guidance 4: Incorporating Clinical Outcome Assessments into Endpoints for Regulatory Decision Making |
Public workshop: Q2 2019 Draft guidance: Q2 2020 Final guidance Q4 2021 |
Public workshop: December 2019 |
|
How FDA will use patient experience data, including with respect to the structured risk-benefit assessment framework |
Public workshop: Q2 2019 Draft guidance: Q2 2020 Final guidance: Q4 2021 |
|
|
Developing and Submitting Proposed Draft Guidance Relating to Patient Experience Data (a.k.a. the “Guidance on Guidances”) |
Draft guidance: Q1 2018 Final guidance: Q4 2019 |
Draft guidance: December 2018 |
[1] See https://www.congress.gov/112/plaws/publ144/PLAW-112publ144.pdf.
[2] See https://www.congress.gov/bill/114th-congress/house-bill/34/text.
[3] See https://www.congress.gov/bill/115th-congress/house-bill/2430.
[4] See https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4486430/.
[5] See https://www.prnewswire.com/news-releases/first-ever-patient-initiated-guidance-for-industry-for-duchenne-muscular-dystrophy-submitted-to-fda-264581331.html.
[6] See https://www.fda.gov/drugs/drug-safety-and-availability/fda-issues-draft-guidance-developing-drugs-duchenne-muscular-dystrophy.
[7] See https://www.fda.gov/regulatory-information/search-fda-guidance-documents/duchenne-muscular-dystrophy-and-related-dystrophinopathies-developing-drugs-treatment-guidance.
[8] See https://www.klobuchar.senate.gov/public/2015/6/klobuchar-wicker-introduce-bipartisan-bill-to-increase-fda-transparency.
[9] See https://www.congress.gov/bill/114th-congress/senate-bill/1597/text?q=%7B%22search%22%3A%5B%22Patient+focused+impact+assessment%22%5D%7D&r=1&s=3.
[10] See https://www.congress.gov/bill/114th-congress/house-bill/34/text.
[11] https://www.congress.gov/bill/114th-congress/house-bill/34/text.
[12] See https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/761083Orig1s000MultidisciplineR.pdf.
[13] Statistics based on a review of the 147 CDER approvals of new molecular entities (NMEs) from January 2018 through September 2020.
[14] See https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/212489Orig1s000MedR.pdf.
[15] See https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761090Orig1s000MultidisciplineR.pdf.
[16] See https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/212273Orig1s000MultidisciplineR.pdf.
[17] See https://www.accessdata.fda.gov/drugsatfda_docs/nda/2020/761158Orig1s000MultidisciplineR.pdf, pages 167 to 168.
[18] See https://www.fda.gov/media/134493/download.
[19] See https://www.fda.gov/drugs/development-approval-process-drugs/external-resources-and-information-related-patients-experience.
[20] See https://www.congress.gov/bill/116th-congress/senate-bill/3385/text?r=1&s=1.
[21] See https://www.govinfo.gov/content/pkg/FR-2016-10-05/html/2016-24100.htm.
[22] See https://www.sciencedaily.com/releases/2015/01/150129170327.htm.
 NICK MANETTO is a principal consultant with Faegre Drinker Consulting where he focuses on a range of federal healthcare policy issues, including those pertaining to the Food and Drug Administration (FDA) and approval of medical products. In his consulting role, he worked on the Patient-Focused Impact Assessment Act and on the 21st Century Cures Act.
NICK MANETTO is a principal consultant with Faegre Drinker Consulting where he focuses on a range of federal healthcare policy issues, including those pertaining to the Food and Drug Administration (FDA) and approval of medical products. In his consulting role, he worked on the Patient-Focused Impact Assessment Act and on the 21st Century Cures Act. LAUREN BLOCH is a director with Faegre Drinker Consulting where her practice focuses on advising patient advocacy organizations on a range of topics, including patient-focused drug development. She works with clients to conduct benefit-risk surveys, organize patient-focused drug development meetings, and respond to regulatory actions.
LAUREN BLOCH is a director with Faegre Drinker Consulting where her practice focuses on advising patient advocacy organizations on a range of topics, including patient-focused drug development. She works with clients to conduct benefit-risk surveys, organize patient-focused drug development meetings, and respond to regulatory actions. ANNIE KENNEDY is the Chief of Policy & Advocacy at the EveryLife Foundation for Rare Diseases where she is focused on improving health outcomes for people living with rare diseases by advancing the development of treatment and diagnostic opportunities for rare disease patients through science-driven public policy.
ANNIE KENNEDY is the Chief of Policy & Advocacy at the EveryLife Foundation for Rare Diseases where she is focused on improving health outcomes for people living with rare diseases by advancing the development of treatment and diagnostic opportunities for rare disease patients through science-driven public policy.
Update Magazine
Winter 2020