
Global Focus: Japan’s Regenerative Medicine Regulatory Pathways: Encouraging Innovation and Patient Access
by Hiroaki Asahara
Since Dr. James Thomson first established a line of human embryonic stem cells 20 years ago, global enthusiasm for the potential of regenerative medicine has led to meaningful progress. Japan is among the countries investing in this emerging technology. On November 21, 2018, the Regenerative Medical Product Section of the Pharmaceutical Affairs and Food Sanitation Council within the Ministry of Health, Labor and Welfare (MHLW) issued an opinion granting Nipro’s Stemirac (for spinal cord injury) conditional market approval as a regenerative medical product.1 Formal conditional market approval was finalized on December 28, 2018.2
This most recent conditional approval of a regenerative medical product in Japan is the culmination of several years of research and regulatory attention in the field. After Dr. Shinya Yamanaka won a Nobel Prize for his work on pluripotent stem cells in 2012, Japanese commitment to regenerative medicine increased significantly. In May 2013, the Japanese legislature passed the Regenerative Medicine Promotion Act and declared its intention to implement necessary reforms to foster the development of regenerative medicine. The government quickly moved this agenda forward by passing the Act on the Safety of Regenerative Medicine (RM Act) and amending the Pharmaceuticals and Medical Devices Act (PMD Act) in November 2013, which became effective in November 2014 (2013 Reform).
The new scheme provided by the 2013 Reform has enabled significant progress in regenerative medicine. This article describes the legislative framework and the paths to market for regenerative medicine, which is unique and controversial and, accordingly, has been watched carefully by stakeholders all over the world.3
The Pharmaceuticals and Medical Devices Act
The first path for regenerative medical products is a “drug” track. In Japan, market approval for drugs and medical devices had been governed by the PMD4 Act, which set forth criteria necessary for market approval issued by the Pharmaceuticals and Medical Devices Agency (PMDA) (i.e., safety and efficacy) and detailed rules required for applicant compliance at both the pre-and post-marketing stage. Under the reform, the category of “Regenerative Medical Product” was newly created.5
As of November 2018, five cases have gone through this path: JACK (skin cells for severe burn) and JACE (chondrocyte tissue for traumatic cartilage deficiency in the knee joint), which were developed by Japan Tissue Engineering; Terumo’s HeartSheet (myoblasts for cardiac insufficiency); TEMCELL HS Injection developed by JCR Pharmaceuticals (mesenchymal stem cells for acute graft versus host disease) and Nipro’s Stemirac.
Importantly, the 2013 Reform authorized expedited market approval for regenerative medical products. Considering the characteristics of regenerative medicine, upon showing safety and presumable clinical benefit, the PMDA can issue time-limited market approval with the condition that the clinical benefit is confirmed during that limited period, which is seven years or less. An application for formal market approval is also mandated within that period. A PMDA officer has likened the criteria for evaluating efficacy under this expedited market approval to those for Accelerated Approval of New Drugs for Serious or Life-Threatening Illnesses in the United States.6 Regenerative medical products approved for marketing, including via expedited conditional market approval, are guaranteed coverage by Japanese social health insurance. This scheme enables patients to access these products with insurance coverage at the stage when they would still be in clinical trials under the conventional market approval scheme (see the chart below).
As of December 2018, Terumo’s HeartSheet and Stemirac obtained this expedited conditional approval. Stemirac was also designated under the Sakigake scheme for accelerating the approval process.7
The practical implication of this expedited approval process is that market approval can be obtained based on a limited number of subjects and surrogate endpoints. For example, HeartSheet received market approval with a single-arm trial of seven subjects evaluated by a surrogate endpoint (LVEF).8 The premarket trial for Stemirac was reported to have 13 subjects.9
Expedited approval is only a preliminary step in the whole product development cycle. In order to obtain expedited approval, it is essential to propose an appropriate R&D design for the entire product cycle, including research and trial plans for the postmarket stage, clearly established indicators for efficacy, and, ultimately, an acceptable threshold for safety and efficacy to support prescribing to patients.10
In the case of HeartSheet, the market approval had a five-year limitation and was conditioned on the conduct of a deeper trial with a clinical endpoint (survival) involving 60 subjects in the product arm and 120 controls who were not treated with the product but shared similar clinical conditions.11 The sponsor applied for an extension of this conditional approval period due to a delay in the trial; a three-year extension, which is the maximum extension allowable under the PMD Act, was approved on November 21, 2018.12
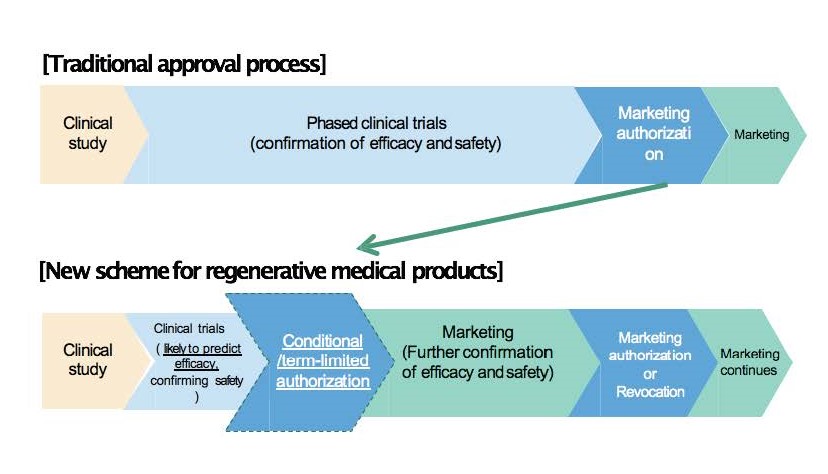
Revised based on a chart by Daisaku Sato (2016) (see note 6)
The Act on the Safety of Regenerative Medicine
Although the first path has successfully accelerated market approval, the PMD Act requirements may prove too burden-some for some regenerative medicine developers, especially for physicians working in early stage development. Thus, a second path—a “clinic” track—is also available. Under this path, regenerative medicine developers, usually physicians, make their products available to patients under the Advanced Medical Care B program of Japan’s Social Health Insurance following clinical research or trials under the RM Act.
In this scenario, even if a medical treatment involves a product that is unapproved or prescribed off-label, Advanced Medical Care B permits partial coverage by social health insurance upon showing of a certain level of efficacy and safety in a clinical trial. This second path also provides a connection to the first path. The data obtained through this process can be used in market approval applications as long as it complies with Good Clinical Practice (GCP) criteria.13 A jaw bone regeneration therapy using bone marrow-derived mesenchymal cells is an example of a therapy that has utilized this second path.14
This path starts from clinical research or trials conducted by physicians involving relatively small number of subjects. This kind of clinical research or trials is not governed by the PMD Act, which applies only to clinical trials conducted to obtain market approval; rather, this research is regulated under the RM Act.15 The RM Act also applies to medical treatment which has been tested in this type of clinical research or trials. In order to assure the safety of regenerative medicine, the RM Act regulates the entire regenerative medicine application—meaning the whole “clinical” track—regardless of whether it is based on medical care, clinical research, or clinical trials, except for clinical trials for market approval. The practical impacts of the RM Act are (i) the requirement to comply with specific criteria for medical care not covered by social health insurance, an area which has not been regulated (the practice of medicine has remained within the professional discretion of physicians except for the regulations attached to Social Health Insurance); (ii) the enactment of regulations for clinical research related to regenerative medicine, which had been regulated by guidelines of MHLW; and (iii) outsourcing is permitted for the manufacture of cell-origin products to approved manufacturers that comply with specific criteria.
The RM Act classifies regenerative medicine subject to the Act into three categories (Class I to III) based on risk, with different procedural requirements for each Class that hospitals or clinics providing regenerative medicine must follow.16 For example, the jaw bone regeneration therapy using bone marrow-derived mesenchymal cells mentioned above was approved as Class II regenerative medicine under the RM Act in July 2015 and designated as Advanced Medical Care B on September 17, 2015.17 Additionally, there is an accelerated path for evaluation of Advanced Medical Care B for treatment classified as Class I regenerative medicine under the RM Act (System for Accelerated Assessments of Advanced Medical Services).18
Conclusion
Following the 2013 Reform, the Japanese government has implemented these two innovative paths for treatment development to strongly support regenerative medicine. This approach is unique to Japan and is considered somewhat controversial in other jurisdictions. Japan’s four-year experience is still not enough to evaluate whether the appropriate balance between safety/efficacy and innovation that the scheme intended to create has been realized. It will be important to continue to keep watch with a skeptical as well as hopeful eye to determine whether Japan’s model for regulating regenerative therapies is successful.
Update Magazine
February/March 2019





- https://www.japantimes.co.jp/news/2018/11/22/national/science-health/health-ministry-panel-gives-conditional-ok-regenerative-medicine-patients-japan-spinal-cord-injuries/#.XAWRY-JNKjOR
- https://headlines.yahoo.co.jp/hl?a=20181228-00000159-kyodonews-soci (Japanese).
- For example, Nature showed skepticism on this legislative framework. See Stem the tide, Nature News (2015), https://www.nature.com/news/stem-the-tide-1.18976.
- Prior to the 2013 Reform, the PMD was called the Pharmaceutical Affairs Act.
- For more details on the definition of this category, see Kentaro Azuma, Regulatory Landscape of Regenerative Medicine in Japan, 1 Current Stem Cell Reports 118—128 (2015).
- Daisaku Sato, Updates on global movement in regulation of Advanced Therapeutics (2016), https://www.pmda.go.jp/files/000209780.pdf.
- For more detail on the Sakigake designation scheme, see Chia-Feng Lu, Japan’s Sakigake Designation System: Promoting Innovation, UPDATE, July/August 2016, at 30
- Yoshiaki Maruyama, Regulation of Regenerative Medicine in Japan, 2017, https://www.pmda.go.jp/files/000219466.pdf.
- https://www.mixonline.jp/Article/tabid/55/artid/65644/Default.aspx (Japanese).
- Kazuhiro Takekita, Regenerative Medical Products: New Regulatory Framework for Conditional and Time-limited Market Approval, 2015, available at https://www.pmda.go.jp/files/000205439.pdf (Japanese).
- Supra note 5.
- Supra note 6.
- Akifumi Matsuyama, Regenerative Medicine: our way for clinical application, 30 Journal of medical law 155-164 (2015) (Japanese).
- https://www.mhlw.go.jp/topics/bukyoku/isei/sensiniryo/kikan02.html (Japanese)
- Initial clinical trials that may eventually lead to seeking market approval (i.e. preliminary step for the “drug” track) may also be conducted under the RM Act. For example, on May 16, 2018, Yoshiki Sawa of Osaka University, who co-developed Terumo’s HeartSheet, obtained approval for a Class I regenerative medicine to commence a clinical trial of heart sheets made of iPS cell. See https://www.nikkei.com/article/DGXMZO30571120W8A510C1MM0000/ (Japanese).
- For more details on the RM Act, see Tobita Morikuni at el., Japan’s challenges of translational regenerative medicine: Act on the safety of regenerative medicine, 4 Regenerative Therapy 78-81 (2016).
- https://saiseiiryo.mhlw.go.jp/published_plan/index/2/2 (Japanese).
- https://www.mhlw.go.jp/file/05-Shingikai-12404000-Hokenkyoku-Iryouka/0000069661.pdf (Japanese).
