
Global Focus
China: Breaking Down the “Great Wall”?
by Xin Tao
Driven by rapid economic growth and the world’s largest rapidly aging population, China’s pharmaceutical market has become the second largest worldwide, only after that of the United States. The market volume is forecasted to grow from US$108bn in 2015 to US$167bn by 2020, representing an annual growth rate of 9.1 percent.1 China also plans to provide basic medical care for its entire population by 2020 and encourages its more affluent population to take out commercial health insurance.2
China, however, has proven to be a difficult market to break into. Historically, regulatory complexities have been a major barrier to global life science companies’ ability to launch innovative drugs in China. For example, the China Food and Drug Administration (CFDA) would not accept clinical trial data developed overseas to support new drug approval. Further, new drugs developed outside of China can only start clinical trials in China after the Phase I clinical trial is completed overseas.3 As a result, of the 433 new drugs approved in developed countries between 2001 and 2016, less than one third are currently marketed in China.4
Another market barrier for global life science companies is the lack of protection against infringing generic drugs. While the current Chinese law requires that a drug applicant disclose whether its product infringes others’ patents, in practice, there are no incentives for drug applicants to do so and also no penalty associated with the failure to disclose. Generic drug products often are approved and sold in the market before a patent holder can initiate any legal action.
To ensure that patients in China have access to innovative products and that pharmaceuticals meet consistent quality standards, in August 2015, the China State Council issued “Opinions on Reforming the Review and Approval System for Drugs and Medical Devices.”5 Since then, CFDA introduced a series of regulatory changes designed to streamline its approval process for new drugs. In June 2017, CFDA was accepted to the International Council for Harmonization (ICH) as a full regulatory member, pledging to transform its regulatory process to be consistent with the international norm.6
Further, for the first time, CFDA plans to adopt a patent linkage system (similar to the Hatch-Waxman system in the U.S.), under which drug applicants are required to make a declaration on patent rights infringement in their applications. The approval of the drug application will be put on hold pending the resolution of a patent dispute. Patent holders will also be able to take the generic drug applicants to courts when the drug application is still pending.
Along with the streamlined approval process and enhanced patent protection comes CFDA’s vigilant overseas inspection program for drug manufacturing. The number of overseas inspections conducted has increased by more than six-fold between 2011 and 2016.7 Major deficiencies identified by CFDA inspectors may lead to suspension of the products’ market authorization.
Faster Market Entry
Recent CFDA regulatory changes that will likely have a substantial bearing on the market entry time of new drugs in China are the CFDA’s acceptance of clinical trial data from studies conducted outside China and the new priority drug review system.
In October 2017, CFDA promulgated Decisions of the China Food and Drug Administration on Issues related to Adjustment of Imported Drug Registration Administration (the Decisions).8 Under the Decisions, with the exception of vaccines, drug applications for multi-regional clinical trials (MRCT) in China are now permitted to launch synchronized Phase I clinical trials both inside and outside China. In other words, this lifts the prior requirement for a new drug to first be approved (or have entered Phase II or III clinical trials) in its home country before clinical trials can be conducted in China. At the conclusion of the MRCTs, the applicants can now directly apply for market authorization without going through the separate registration process for import drugs. The previous requirements that all import drugs must be approved overseas before they can be authorized in China are also abolished for new chemical drugs and innovative biologics. Global companies should reassess their clinical trial strategy in light of these changes, as these changes will permit the submission of drug applications in China in parallel with applications in the U.S.
In 2016, CFDA promulgated a new priority review system to foster innovation.9 For new drugs, drug applicants may apply for a priority review status when the products meet one of the following criteria by submitting applications to CFDA’s Center for Drug Evaluation (CDE):
- Innovative drugs not approved anywhere in the world
- Innovative drugs where there is a plan to be manufactured in China
- Innovative drugs that are under parallel clinical trial applications in the U.S. and the EU
Other drugs, including drugs for HIV, tuberculosis, viral hepatitis, rare diseases, malignant tumors, pediatric uses, and diseases afflicting the elderly, may also qualify for priority review under limited circumstances. CDE will review the priority review status applications. Once the application is approved, the drug applicant will have additional channels of communication with CDE before the formal submission of an application to commence clinical trials in China. Discussion during these presubmission meetings can include topics such as the sufficiency of the current research data to support a Phase I clinical trial exemption.
These changes have the potential of greatly shortening the amount of review time. Indeed, in March 2017, seven months after a global company submitted its application for Tagrisso (osimertinib) for the treatment of adult patients with lung cancer, CFDA granted marketing authorization. In its press release, the company notes that the rapid review and approval is “recognition by the CFDA of the accelerated reform of China’s regulatory framework to benefit Chinese patients.”10
Enhanced Protection for Patent Holders
Another encouraging development is the strengthening of intellectual property protection for innovative drugs. In May 2017, CFDA issued a policy document titled “Policies Regarding the Promotion and Protection of Innovators’ Rights in Drugs and Medical Devices (draft for public comments).”11 Key elements of this new policy include the new drug patent linkage system and clinical trial data exclusivity.
Under the proposed patent linkage system, the drug applicant is required to disclose whether the product infringes others’ patents. In the event the applicant decides to challenge the patent, a notification is required to be sent to the patent holders within 20 days of the application submission. If the patent holder views the applicant’s product as an infringing product, a complaint should be filed with the courts within 20 days of receiving the notice from the drug applicant. Patent holders can also inform CFDA of any infringing drug application. CFDA in turn, can decide whether it should set up a 24-month wait period, during which the drug application cannot be approved pending the resolution of the dispute. However, if no settlement or decision is made after 24 months, CFDA can approve the product.
The new policy also expands the scope of clinical trial data exclusivity, which prohibits other applicants from using these data in their drug applications, from only new chemical entity drugs to cover all innovative drugs, orphan drugs, pediatric drugs, and certain generic drugs. We summarize the periods of data exclusivity for different product categories in the table below:
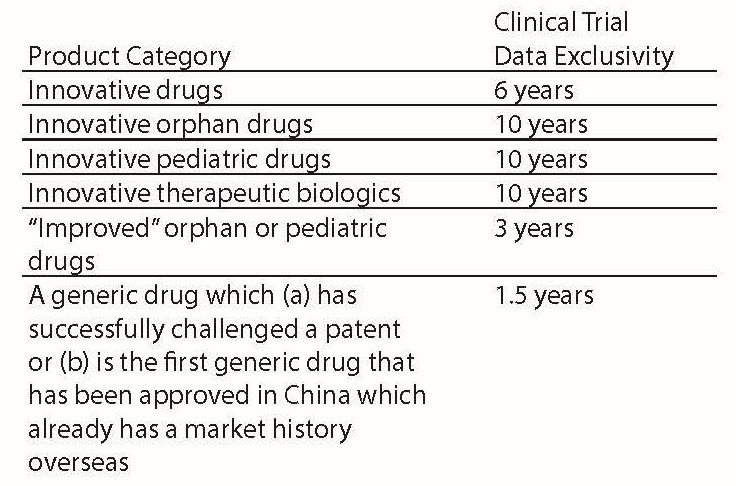
Also, notably, the data exclusivity can only be triggered when the drug applicant explicitly applies for it when applying for market authorization. Further, to encourage global companies to bring more innovative drugs to China, the above data exclusivity periods only apply when the drug application is filed within one year of the new drug’s market approval in U.S., EU, or Japan. If the application in China is filed more than one year after the approval is issued in these developed countries, the data exclusivity period will be deducted accordingly with any time delay exceeding one year.
CFDA Expands Overseas Inspection Program
It should also be noted that in addition to regulatory changes that help break down the market barriers for global companies, CFDA also expanded its inspection program for pharmaceuticals manufactured overseas. Failing a CFDA inspection may carry serious legal significance and can lead to immediate suspension of the products’ market authorization in China. Companies currently selling into China should get familiarized with the Chinese current good manufacturing practice (cGMP) standards and not assume their quality system will always pass muster. It is also helpful to monitor the CFDA inspection observations and identify the trends.
According to CFDA’s annual report of inspections in 2016 (the most recent year that the annual report is available), a total of 49 CFDA overseas inspections took place in 19 countries. At the conclusion of these inspections, a total of 117 deficiencies were identified, of which there were three critical deficiencies and 18 major deficiencies. According to CFDA, the deficiencies observed were mainly associated with Quality Control and Quality Assurance, Material System and Change Management. The critical deficiencies were due to the inconsistency of production process and data integrity issues. These findings led to the suspension of market authorization of four drugs manufactured overseas in Austria, Japan, Italy, and India.12
Conclusion
The recent CFDA reform, which is designed to provide Chinese patients with better access to innovative drugs, will also benefit global companies by breaking down the regulatory barriers. The changes provide tremendous opportunities for fostering new innovations and improving access within China. Further, as CFDA continues to modernize its regulatory regime, we also observe more enforcement actions against global companies for failing to comply with Chinese cGMP standards. As global companies continue to develop new positioning strategies for doing business in the marketplace, they’ll also find new opportunities to leverage the sweeping regulatory and policy changes that continue to evolve in China.
- International Trade Administration, 2016 Top Markets Report Pharmaceuticals Country Case Study.
- 《国务院办公厅关于印发全国医疗卫生服务体系规划纲要(2015-2020)的通知》
- At the time of this publication, the Chinese government proposes to merge CFDA into a national market supervision administration and the drug regulation function will be diverted to a new agency. To avoid any confusion, we continue to use CFDA throughout this article.
- 新华网,“中国药品审批改革提速 国外新药进入中国时间将缩短,” available at http://www.xinhuanet.com/fortune/2017-10/10/c_1121776626.htm.
- 《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号).
- ICH Assembly, Montreal, Canada, May/June 2017, available at: http://www.ich.org/ichnews/press-releases/view/article/ich-assembly-montreal-canada-mayjune-2017.html
- CFDA, Annual Report of Drug Inspection 2016, available at: http://eng.sfda.gov.cn/WS03/CL0757/173386.html.
- 《国家食品药品监督管理总局关于调整进口药品注册管理有关事项的决定》(国家食品药品监督管理总局令第35号).
- 《总局关于鼓励药品创新实行优先审评审批的意见 》(食药监药化管〔2017〕126号).
- Press Release, “Tagrisso approved in China as first-in-class treatment for EGFR T790M mutation-positive metastatic non-small cell lung cancer,” available at: https://www.astrazeneca.com/media-centre/press-releases/2017/tagrisso-approved-in-china-as-first-in-class-treatment-for-egfr-t790m-mutation-positive-metastatic-non-small-cell-lung-cancer-27032017.html.
- 《总局关于征求《关于鼓励药品医疗器械创新保护创新者权益的相关政策(征求意见稿)》(2017年第55号).
- 《关于停止进口脑蛋白水解物注射液等4个药品的公告》(2016年第13号).
Update Magazine
March/April 2018








