Factors to Consider Before Submitting a de novo Request
by Michele L. Buenafe
de novo requests are becoming an increasingly important premarket pathway option for medical devices, particularly those involving novel technologies. First established under the Food and Drug Administration Modernization Act of 1997, the de novo reclassification process was intended as a less burdensome premarket pathway for novel devices that had not previously been classified by FDA and, thus, were considered Class III by default. However, this process was long viewed by both industry and FDA as a less desirable premarket review option. The original de novo process was cumbersome and inefficient, requiring a sponsor to first submit a 510(k) and have it rejected by FDA as not substantially equivalent (NSE) before submitting a de novo request. Consequently, the review times for de novo requests were as long as, and often exceeded, those for premarket approval (PMA) applications. Because the de novo process was so burdensome, FDA reviewers in the Center for Devices and Radiological Health (CDRH) were often more lenient in the use of 510(k)s, allowing some novel or modified devices to be cleared through the 510(k) process by stretching the bounds of substantial equivalence.
 To address FDA and industry concerns, Congress streamlined the de novo process under the Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA), which allows sponsors to submit “direct” de novo requests by skipping the 510(k) NSE step. Since the enactment of FDASIA, FDA has increased its emphasis on the de novo process, encouraging, and in some cases requiring, medical device manufacturers to employ this underutilized pathway for novel or significantly modified devices. Although the de novo review process has greatly improved, challenges remain as FDA and industry work through the kinks of a relatively new premarket approach.
To address FDA and industry concerns, Congress streamlined the de novo process under the Food and Drug Administration Safety and Innovation Act of 2012 (FDASIA), which allows sponsors to submit “direct” de novo requests by skipping the 510(k) NSE step. Since the enactment of FDASIA, FDA has increased its emphasis on the de novo process, encouraging, and in some cases requiring, medical device manufacturers to employ this underutilized pathway for novel or significantly modified devices. Although the de novo review process has greatly improved, challenges remain as FDA and industry work through the kinks of a relatively new premarket approach.
This article discusses issues that device manufacturers should consider when evaluating whether to submit a de novo request.
1. Your Choices May Be Limited
With the direct de novo option now on the table, CDRH’s review divisions appear committed to increasing the utilization of the de novo review process and making it work. To that end, FDA has not only been encouraging industry to submit de novo requests, but also has been more stringent in the use of the 510(k) pathway. For example, FDA’s 2014 final guidance on Evaluating Substantial Equivalence in Premarket Notifications1 discusses restrictions on when multiple predicates can be used and the unacceptability of the use of “split predicates” in 510(k)s. This final guidance also addresses the two key factors that could trigger an NSE determination—a new intended use and new or different technological characteristics that raise new questions of safety or effectiveness. The final guidance provides examples of when each factor could be triggered and result in an NSE determination, putting manufacturers on notice of the types of changes that may not be appropriate for the 510(k) pathway.
2. FDA Review Times are Still Long
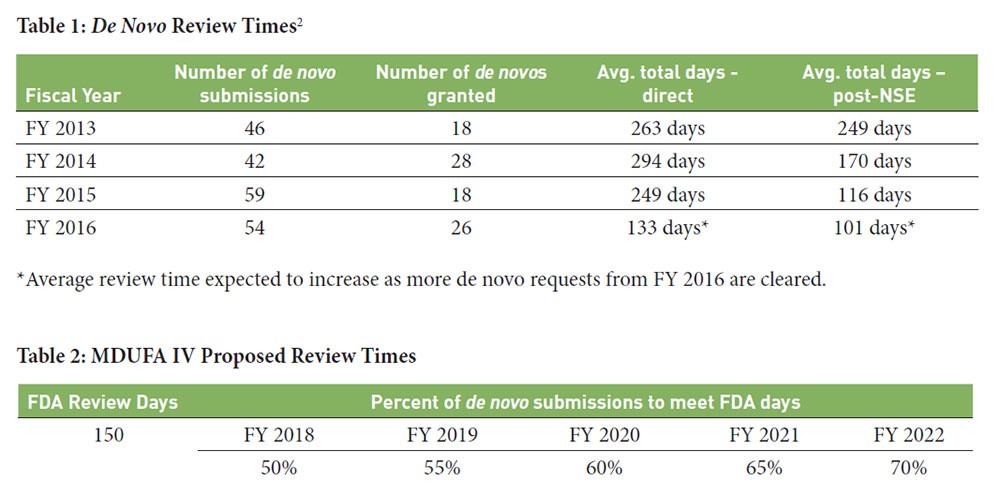
Although the Federal Food, Drug and Cosmetic Act (FDCA) provides that FDA has 120 days to review de novo requests, FDA review times have generally been longer. CDRH does not currently have dedicated resources to handle de novo requests. Although review times have generally improved since the enactment of FDASIA and the availability of the direct de novo process, the average review time is still more than twice the statutory time, as shown in Table 1.
Under the proposed outline for the 2017 Medical Device User Fee Amendments (MDUFA IV), FDA has agreed to progress towards issuing a MDUFA decision for de novo requests within 150 days, as shown below in Table 2.3
Although this is still outside of the current statutory requirement for review within 120 days, it’s important to note that currently only about 40% of de novo submissions are reviewed in 150 days. Thus, the MDUFA IV commitments are not inconsequential.
3. The de novo Process is Not Limited to Low Risk Devices
In the past, the majority of de novo submissions were for devices that were clearly low risk. More recently granted de novo requests, however, include devices that are pushing towards the higher end of the Class II risk spectrum. For example the de novo request, DEN150049, was granted in September 2016 for a neurovascular mechanical thrombectomy device for acute ischemic stroke, which FDA reclassified as Class II.4 This new generic device type is “delivered into the neurovasculature with an endovascular approach, mechanically removes thrombus from the body, and restores blood flow in the neurovasculature,” and is “used in the treatment of acute ischemic stroke to improve clinical outcomes.”5
Another example is the de novo request, DEN150011, granted in October 2015 for a high intensity ultrasound system for prostate tissue ablation, which was reclassified as Class II.6 This new generic device type is defined as “a prescription device that transmits high intensity therapeutic ultrasound (HITU) energy into the prostate to thermally ablate a defined, targeted volume of tissue, performed under imaging guidance.”7
It’s important to note, however, that pushing the envelope for de novo submissions can result in a more intense and lengthy FDA review. For example, in the past year FDA has subjected two de novo requests to advisory panel review, including DEN150035 for a lysosomal storage disorder newborn screening test system8 (granted February 3, 20179), and DEN160043 for a cerebral protection system for use in patients undergoing transcatheter aortic valve replacement (meeting held February 23, 2017, de novo request still pending).10
4. The Standard of Review and Data Requirements
FDA may grant a de novo request if the sponsor can establish that its device is appropriate for classification into Class I or Class II per Section 513(a)(1) of the FDCA. This requires the sponsor to demonstrate the safety and efficacy of the device can be reasonably assured by general controls (for Class I) or by general and special controls (for Class II). These standards, however, were originally drafted for use in classifying or reclassifying an entire device type. FDA’s classification/reclassification processes are generally much larger efforts, involving input from all stakeholders, public meetings, notice-and-comment rulemaking, and, in many cases, a classification panel. Utilizing these classification standards in the context of a 120-day premarket review process for a single device has presented challenges and FDA has yet to provide clear guidance for sponsors on how to meet this standard of review or what level of data can be used to demonstrate that the standard has been met.
FDA’s initial volley on this issue came in the form of its 2014 draft guidance, de novo Classification Process (Evaluation of Automatic Class III Designation).11 This draft guidance states that sponsors should include in their de novo submissions “a summary of all performance and clinical testing data that provide a reasonable assurance of the safety and effectiveness of your specific device and that demonstrate that general controls or general and special controls are sufficient to provide reasonable assurance of safety and effectiveness.”12 Thus, the draft guidance suggests that, unlike 510(k)s where sponsors provide data to show substantial equivalence to a predicate device, the data requirements for de novo submissions more closely resemble those for PMAs, in which sponsors are required to demonstrate reasonable assurance that the subject device is safe and effective for its intended use.13 On the other hand, the de novo pathway is intended to be a less burdensome alternative to the PMA route; thus, the data requirements for safety and effectiveness would presumably be less than for a PMA. How much less, however, is not yet clearly defined.
In addition to the more general requirement of establishing that there is reasonable assurance that the subject device is safe and effective, de novo sponsors also must identify all known risks associated with the device and then identify how such risks can be mitigated through existing general controls or proposed special controls. As described further below, the proposed special controls are a key part of establishing requirements for follow-on devices.
5. Follow-on Competitors and the Importance of Special Controls
A device cleared through the de novo process is immediately available as a predicate for follow-on devices, which may be cleared through the 510(k) process. Thus, the standard of review for follow-on devices is different, and arguably less burdensome (substantial equivalence vs. demonstration that general/special controls provide reasonable assurance of safety and efficacy). Moreover, the 510(k) premarket notification process is a well-worn process with which both FDA and industry are intimately familiar, and FDA reviewers will not be faced with the same uncertainties that they face when reviewing an entirely novel device.
Companies seeking to mitigate the risk of follow-on competitors should carefully consider what special controls to propose in their de novo submissions. Per FDA’s 2014 draft guidance on the de novo Classification Process, FDA requires that a de novo request that proposes reclassifying a device as Class II include proposed special controls. Although follow-on devices will be able to utilize the 510(k) review process, they will still need to comply with the relevant special controls. de novo sponsors should, therefore, consider what premarket testing would be appropriate for the new generic device type, which may include, for example, clinical testing, non-clinical performance data, animal testing, and software validation. However, any premarket testing requirements proposed as special controls in the de novo submission must be consistent with the testing that is included for the subject device.
6. User Fees Under MDUFA IV Will Be Significant
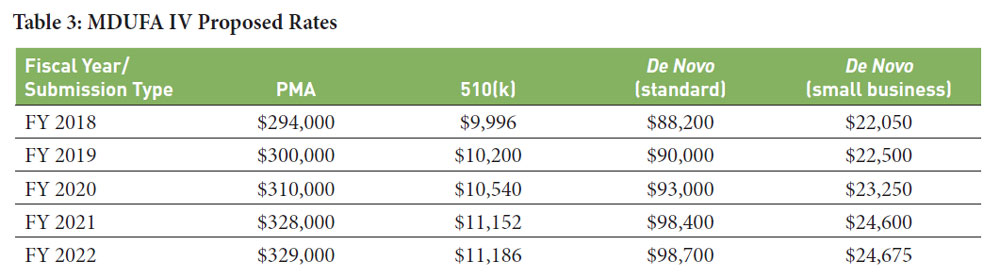
Although de novo requests are not currently subject to user fees, this is almost certain to change under MDUFA IV. Moreover, the user fees for de novo submissions will be significantly more than those for 510(k)s. FDA’s proposal for MDUFA IV recommends that user fees for de novo requests be priced at 30% of those for PMAs.14 For small businesses, the de novo user fee would be 25% of the standard de novo rate.15 User fees for 510(k)s also are increasing from 2.0% of the PMA rate to 3.4%, but even with this increase, the 510(k) fees will be substantially lower than the de novo fees.16 Any company intending to submit a de novo request in 2017 should consider, when planning its submission timeline, that the expected increase in user fees will take effect October 1, 2017 (the first day of FDA’s 2018 fiscal year), assuming that MDUFA IV is enacted into legislation in a timely manner.
Table 3 summarizes the proposed rates for PMAs, 510(k)s, and de novo requests under MDUFA IV.
- FDA, The 510(k) Program: Evaluating Substantial Equivalence in Premarket Notifications [510(k)], Guidance for Industry and Food and Drug Administration Staff (July 28, 2014), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm284443.pdf.
- FDA, MDUFA III Performance Report (Dec. 9, 2016), https://www.fda.gov/downloads/ForIndustry/UserFees/MedicalDeviceUserFee/UCM532387.pdf.
- FDA, MDUFA Performance Goals and Procedures, Fiscal Years 2018 Through 2022 (Dec. 2, 2016), https://www.fda.gov/downloads/ForIndustry/UserFees/MedicalDeviceUserFee/UCM535548.pdf.
- FDA Reclassification Order for DEN150049 (Sept. 2, 2016), http://www.accessdata.fda.gov/cdrh_docs/pdf15/DEN150049.pdf.
- Id.
- FDA Reclassification Order for DEN150011 (Oct. 9, 2015), http://www.accessdata.fda.gov/cdrh_docs/pdf15/DEN150011.pdf.
- Id.
- FDA Executive Summary, Clinical Chemistry and Clinical Toxicology Devices Panel, DEN150035 (Aug. 10, 2016), https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/MedicalDevices/MedicalDevicesAdvisoryCommittee/ClinicalChemistryandClinicalToxicologyDevicesPanel/UCM515322.pdf.
- FDA Reclassification Order for DEN150035 (Feb. 3, 2017), http://www.accessdata.fda.gov/cdrh_docs/pdf15/DEN150035.pdf.
- FDA Executive Summary, Circulatory System Devices Panel Meeting, DEN160043 (Feb. 23, 2017), https://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/MedicalDevices/MedicalDevicesAdvisoryCommittee/CirculatorySystemDevicesPanel/UCM542415.pdf.
- FDA, de novo Classification Process (Evaluation of Automatic Class III Designation), Draft Guidance for Industry and Food and Drug Administration Staff (Aug. 14, 2014), https://www.fda.gov/ucm/groups/fdagov-public/@fdagov-meddev-gen/documents/document/ucm273903.pdf.
- Id. at 15 (emphasis added).
- See 21 U.S.C. § 360e(d)(1)-(2).
- FDA, Summary of Draft Recommended Changes to Statutory Language for MDUFA IV (Oct. 25, 2016), http://www.fda.gov/downloads/ForIndustry/UserFees/MedicalDeviceUserFee/UCM526532.pdf.
- Id.
- Id.
Update Magazine
March/April 2017